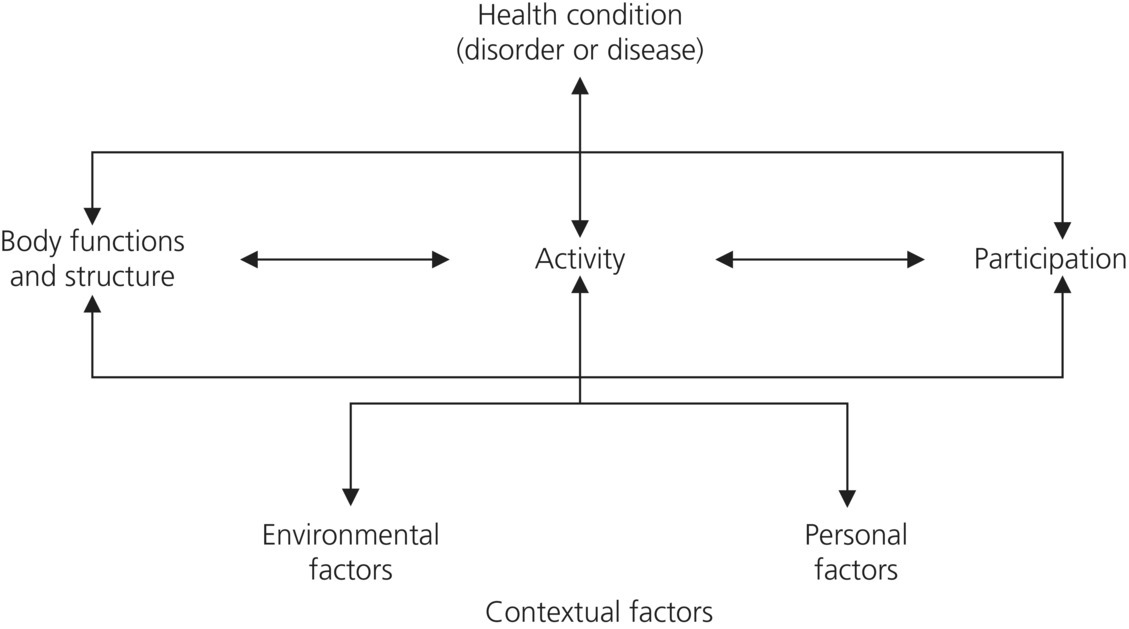
CHAPTER 1 It is estimated that 10 million people in the United Kingdom live with some form of neurological condition that impacts on their everyday lives (Department of Health [DoH], 2005). Neurological conditions account for one in five emergency hospital admissions, one in eight general practice consultations and a high proportion of severe and progressive disability in the population (Association of British Neurologists, 2003). As many as 350 000 people in the United Kingdom need help with activities of daily living because of a neurological condition and 850 000 people care for someone with a neurological condition (DoH, 2005). Due to their devastating impact and their generally progressive nature, neurological conditions are considered as long-term affecting individuals throughout their life span. Occupational therapy is defined as ‘a client-centred health profession concerned with promoting health and well-being through occupation enabling people to participate in everyday life’ (World Federation of Occupational Therapists, 2011). Occupational therapy practice focuses on enabling individuals to modify and adapt elements of their roles, occupations or environments to support occupational participation in response to changes within their lives. Occupational therapists have a key role to play in supporting people living with a long-term neurological condition to manage a life of unpredictability and uncertainty. This requires a complex combination of knowledge and skills to address the physical, psychological, cognitive and emotional needs of people together with a broad range of assessments and interventions. The DoH (2005) describes ‘long-term neurological conditions’ as a range of conditions affecting the brain or spinal cord which occur through a variety of mechanisms which include the following: This book specifically focuses on the following progressive neurological conditions: Whilst there is an abundance of literature relating to each of these medical conditions the primary aim of this book is to place this knowledge and understanding within the context of occupational therapy practice. In order to fully understand the holistic needs of their clients occupational therapists are required to develop knowledge of the underlying pathology of each of these neurological conditions. However this understanding from a medical perspective should not be assumed to represent a medical model of care with an emphasis on symptomatic management. Throughout this book the focus is on delivering person-centred models of practice which support the complexity of the needs of people with neurological conditions from an occupational perspective. The International Classification of Functioning, Disability and Health (ICF) offers a conceptual basis for the definition and measurement of health and disability (World Health Organisation [WHO], 2002). Developed within a biopsychosocial model, ICF views disability and functioning as outcomes of interactions between health conditions (diseases, disorders and injuries) and contextual factors, as shown in Figure 1.1. Amongst contextual factors are external environmental factors (e.g., social attitudes, architectural characteristics, legal and social structures) and internal factors which include gender, age, coping styles, social background, past and current experience, character and other factors that influence how disability is experienced by the individual (WHO, 2002). Figure 1.1 Model of disability that is the basis for ICF. (Source: WHO, 2002, p. 9. Reproduced with permission of World Health Organisation.) Within this framework ICF defines three levels of human functioning: functioning at the level of body or body part (impairment), the whole person (activity limitations) and the whole person in a social context (participation restrictions). The formal definitions of these components of ICF are provided in Box 1.1 (WHO, 2002). The remainder of this chapter presents each of the four neurological conditions in relation to body functions, body structures and impairments, highlighting the differences and similarities of each condition. Subsequent chapters explore the wider implications for activity and participation. HD is a rare disease, affecting an estimated 7–10 people per 100 000 or somewhere in the region of between 4200 and 6000 people in the United Kingdom (Quarrell, 2008). The onset of the disease is insidious and the age of onset depends on a number of different factors. Most people develop the condition between the ages of 30 and 50 years, but the disease can appear in all age groups (Nance et al., 2013). The HD gene is dominant, which means that each child of a parent with HD has a 50% chance of inheriting the disease and is said to be ‘at-risk’. Males and females have the same risk of inheriting the disease. HD occurs in all races (Nance et al., 2013). There is currently no cure or treatment which can halt, slow or reverse the progression of the disease (Nance et al., 2013) and people with HD tend to die, on average, between 15 and 16 years after the onset of symptoms (Quarrell, 2008). People don’t die from HD itself, but they die from complications such as choking, heart failure, and infection or aspiration pneumonia (Nance et al., 2013). HD is a hereditary neurodegenerative genetic disorder caused by an expansion of a repeating CAG triplet series in the huntingtin gene on chromosome 4, which results in a protein with an abnormally long polyglutamine sequence (Nance et al., 2013). HD causes cells in the brain to die, specifically the caudate and the putamen and, as the disease progresses, the cerebral cortex. These organic changes lead to cognitive, motor and psychiatric changes that have a devastating impact on the individual. As the brain cells die, a person with HD becomes less able to control their movements, recall events, make decisions and control their emotions (Nance et al., 2013). Symptoms may include minor involuntary movements, subtle loss of coordination, difficulty thinking through complex problems, depression, irritability, or disinhibition (Nance et al., 2013). Early symptoms of the disease often include subtle cognitive changes including the following: Chorea may be prominent, and people with HD have increasing difficulty with voluntary motor tasks. There may be issues with swallowing, balance, falls and weight loss. Problem solving becomes more difficult due to difficulties sequencing, organising or prioritising information (Nance et al., 2013). The initial physical symptoms will gradually develop into more obvious involuntary movements such as jerking and twitching of the head, neck and arms and legs. These movements may interfere with walking, speaking and swallowing. People at this stage of HD often stagger when they walk and their speech may become slurred. They may have increasing difficulty working or managing a household, but they can still deal with most activities of daily living (Nance et al., 2013). Chorea may be severe, or be replaced by rigidity, dystonia and bradykinesia. Although they are unable to speak in the end stages, it is important to note that people with HD retain a level of comprehension (Nance et al., 2013). People in these stages of HD can no longer manage the activities of daily living and usually require professional nursing care. Difficulties with swallowing and weight loss are common (Nance et al., 2013). More than 90% of people with HD have chorea. It is characterised by ‘involuntary movements which are often sudden, irregular and purposeless or semi-purposeful. The movements are often more prominent in the extremities early in the disease, but progress to include facial grimacing, eyelid elevation, neck, shoulder, trunk, and leg movements as the disease progresses’ (Nance et al., 2013). Characterised by ‘a repetitive, abnormal pattern of muscle contraction which is frequently associated with a twisting quality’ (Nance et al., 2013). ‘Slowness of movement can include loss of facial expressivity, absence of arm swing, rapid alternating movements and gait slowness’ (Nance et al., 2013). ‘are sudden brief, intermittent movements, gestures or vocalisations which can occur with HD. Respiratory and vocal tics can produce sniffs, grunts, moans or coughs’ (Nance et al., 2013). Genetic testing in HD can serve two purposes: as a diagnostic tool and as a predictive test to identify level of risk. Genetic testing involves the examination of an individual’s DNA, which is obtained from a blood sample. DNA molecules consist of four bases, known as A (adenine), T (thymine), G (guanine) and C (cytosine). The gene that causes HD is called the HD gene, and within it there is a region in which a sequence of the three bases (CAG) is repeated many times. For individuals with HD, the CAG sequence has increased (expanded) into a range that is abnormal. Testing is done in a specialised laboratory to determine the number of CAG repeats in both copies of the HD gene (Huntington’s Disease Association, 2012). An HD gene expansion is passed on in families and children of a parent with this expansion have a 50% chance of developing the disease. Predictive testing is a process whereby an individual at risk of the disease can discover whether or not they have inherited the expanded HD gene, and will go on to develop HD. A ‘gene negative’ result is where the number of CAG repeats is 26 or less. The individual will not go on to develop the HD and their children will not be at increased risk either (Huntington’s Disease Association, 2012). An intermediate result is a result where the number of CAG repeats is between 27 and 35. This means that the individual will not go on to develop HD but, in some cases, may pass on an expansion to their children because the CAG repeat can be unstable when passed from one generation to the next. This can mean that sometimes children will be at higher risk for developing HD (Huntington’s Disease Association, 2012). A reduced penetrance result is one where the number of CAG repeats is between 36 and 39. An individual with a result in this range may not develop any symptoms of HD; however, this result also means that the next generation may be at risk of inheriting a larger expansion as it would also be unstable (Huntington’s Disease Association, 2012). A full penetrance or ‘gene positive’ is a result where the number of CAG repeats is 40 or more. The individual with this result will always go on to develop HD at some point in the future. The result does not give information on the age of onset of symptoms (Huntington’s Disease Association, 2012).
Introduction
1.1 Economic impact of long-term neurological conditions
1.2 Definition of long-term neurological conditions
1.3 International Classification of Functioning, Disability and Health
1.4 Huntington’s disease
1.4.1 Body functions
1.4.2 Body structures
1.4.3 Stages of HD
Early stage
Middle stage
Late stage
1.4.4 Impairments
1.4.5 Diagnosing HD
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


