CHAPTER 14 Laura J. Julian1 and Christopher M. Filley2,3 1 University of California, San Francisco, San Francisco, CA, USA 2 University of Colorado School of Medicine, Denver, CO, USA 3 Denver VA Medical Center, Denver, CO, USA The intersection of rheumatology and neurology, particularly with respect to central nervous system (CNS) and cognitive manifestations, is undoubtedly complex and presents unique challenges for each specialty. For neurologists who most often see these patients in consultation, neurologic complications of rheumatic disease present the challenge of understanding and managing the complex interactions of the immune system with the nervous system. For rheumatologists, CNS complications of rheumatic diseases present some of the most difficult clinical problems as well as the most potentially disabling complications for their patients. The scope of this chapter will be limited to conditions that fall within the discipline of rheumatology and that can cause dementia or significant cognitive impairment. Systemic lupus erythematosus (SLE) is the rheumatologic disease whose neuropsychiatric manifestations have received the most attention from both clinicians and researchers, and a sizeable proportion of our discussion will be focused on this disorder. In addition, we will also discuss other rheumatic disorders with the propensity for producing neuropsychiatric sequelae, including the antiphospholipid antibody syndrome (APS), Sjögren’s syndrome, the vasculitides, systemic sclerosis (scleroderma), sarcoidosis, and celiac disease. The clinical information about CNS manifestations of rheumatologic disease is variable and in many cases sparse, reliant solely on case reports and case series. The CNS complications of these conditions can result in a range of cognitive effects, from relatively mild cognitive impairment to severe dementia. In the majority of these conditions, the cognitive loss is characterized by a loss in function compared to a previous level and has the potential to impair functioning in social, occupational, and educational domains while not meeting the severity level of dementia. For the purposes of this chapter, we define cognitive dysfunction conventionally as the presence of significant deficits in any cognitive domain, which can include learning and memory, executive function, language, visuospatial function, information processing speed, and simple and complex attention. In the practice of rheumatology as well as neurology, there is a high likelihood of encountering patients with milder degrees of cognitive dysfunction that are disconcerting to the patient and disruptive to daily functioning, yet not always detectable in the general clinical encounter, particularly when other neurologic functions (e.g., motor, sensory) appear to be unaffected. Therefore, we emphasize in this chapter the benefit of employing a multidisciplinary approach to patient evaluation and treatment, specifically through the use of neuropsychological assessment. Neuropsychological testing can facilitate quantification of cognitive deficits that cannot be as thoroughly assessed in the clinical encounter, serve to track the course of cognitive functioning, and help determine the efficacy or toxicity of specific treatments on cognition. We recommend the use of neuropsychology as an integrated component of the clinical evaluation not only because subtle deficits may escape recognition in the clinical encounter but also because subjective complaints of cognitive decline by patients are not always objectively confirmed. This discordance among patient reports of cognitive dysfunction and performance on neuropsychological testing may stem from several sources, including the influence of psychological distress (e.g., depression) [1], the impact of fatigue, and the insensitivity of cognitive screening measures to more subtle alterations in cognition [2]. The neurologist typically becomes involved in the care of a rheumatologic patient under two primary circumstances. The first is in the context of a known and previously diagnosed rheumatologic condition after the onset and/or progression of CNS or peripheral nervous system involvement. The second, and more challenging scenario, is when the initial presentation of the patient to the neurologist occurs before the underlying rheumatologic condition is diagnosed. In this latter situation, there is no typical presentation, and the spectrum of potential neurologic presenting complaints is broad, ranging from headaches and malaise to stroke, transverse myelitis, peripheral neuropathy, and cognitive impairment. The majority of rheumatologic diagnoses are reached through a careful characterization of the patient’s clinical syndrome using the history, physical examination, and laboratory testing and with the exclusion of nonrheumatologic conditions. The use of laboratory studies for the diagnosis of rheumatologic conditions has become increasingly refined and remains a key feature of the diagnostic process. There are few if any rheumatologic conditions that can be definitively diagnosed through laboratory testing, however, and it remains important to understand the limitations of diagnostic tests and the potential problems with interpreting specific laboratory measures. For example, an essential feature of any rheumatologic panel is testing for antinuclear antibodies (ANA). Whereas this is a reasonable approach, as these autoantibodies are considered a hallmark of systemic autoimmune disease, it is important to consider that the sensitivity of a positive ANA for a specific autoimmune disease varies widely, from 48% in Sjögren’s syndrome to 93% in SLE, and that many people in the general population with a positive ANA do not have an identifiable systemic disease [3]. Another example is the testing of antiphospholipid antibodies, which should be considered for the assessment of neurologic complications of SLE, primary APS, and Sneddon’s syndrome. Whereas testing for such antibodies as anticardiolipin antibodies (AcL), beta-2-glycoprotein 1 antibodies, and the lupus anticoagulant (LAC) can provide clues to the pathogenesis of clotting disorders and cerebrovascular (CV) disease, only repeat testing at least 12 weeks after the initial testing can be used to make a definitive diagnosis of APS [4]. Consideration of a rheumatic condition in a patient presenting with a neurologic complaint requires a thorough evaluation of extraneurologic symptoms and manifestations as well as those involving the nervous system. For example, evaluation of constitutional symptoms, organ involvement, joint manifestations, skin rashes, and hematologic abnormalities can all be helpful. Table 14.1 lists the conditions reviewed in this chapter along with the range of neurologic features, primary nonneurologic features, and potential neurobehavioral sequelae. We will briefly describe each condition, discuss the major cognitive and/ or neurological manifestations, highlight specific diagnostic approaches and findings (including neuroimaging), and briefly discuss aspects of prognosis as well as interventions in clinical practice. To illustrate the complexity of diagnosis and treatment that may characterize rheumatologic patients with neurologic manifestations, the following case is presented. Table 14.1 Rheumatologic and related dementias and neurological characteristics. A 75-year-old right-handed man was referred for 1 year of progressive cognitive decline. He had been noted to develop apathy, inattention, and poor memory, followed by gait disorder and incontinence of bladder and bowel. Brain magnetic resonance imaging (MRI) showed scattered ischemic white matter (WM) changes, and electroencephalography (EEG) showed frontal intermittent rhythmic delta activity. Three months after onset, cerebrospinal fluid (CSF) examination showed clear fluid with glucose 41 mg/dL, protein 117 mg/dL, 33 white blood cells (WBCs) per μL with lymphocytic predominance, and 14 red blood cells (RBCs) per μL. In addition, the CSF had evidence of low amyloid β42 (509 pg/mL) and high tau (709 pg/mL), resulting in an amyloid β42/tau ratio interpreted as consistent with Alzheimer’s disease (AD). EEG at that time showed bilateral periodic lateralized epileptiform discharges. Other laboratory tests for reversible causes of dementia were unremarkable. Despite the inflammatory CSF, a diagnosis of probable AD was made. Cognition and neurologic status continued to worsen, and 5 months after disease onset, the score on the Mini-Mental State Examination (MMSE) [5] was 14, with poor orientation and attention and 1/3 word recall. At 8 months, the MMSE score was 9, with marked apathy and 0/3 word recall. At 10 months, he was nearly mute, had muscle rigidity, and could neither stand nor walk. The patient was then referred for a second opinion. When examined at the University of Colorado Hospital Neurobehavior Clinic 1 year after disease onset, the patient was profoundly apathetic and the MMSE could not be administered. He had diffuse rigidity, the inability to stand or walk, and bilateral grasp reflexes. The CSF pleocytosis documented months before was recognized, and he was emergently admitted to the hospital for thorough evaluation. MRI scan showed ventricular enlargement and meningeal inflammation (Figure 14.1). CSF examination again showed lymphocytic pleocytosis, this time with 10 WBCs/μL, and elevated protein of 108 mg/dL; glucose was 45 mg/dL. An extensive workup for infectious disease and neoplasia was unrevealing, and the ANA was negative, but the perinuclear antineutrophil cytoplasmic antibody (P-ANCA) titer was positive at 1:320 with negative cytoplasmic ANCA (C-ANCA). Chest roentgenogram and pulmonary computed tomography (CT) showed mediastinal adenopathy and right lower lobe consolidation, and diagnostic possibilities including Wegener’s granulomatosis and sarcoidosis were considered. Bronchoscopy was felt to be of low yield, however, and was not performed. The decision was then made to obtain brain biopsy, and tissue taken from the right frontal lobe and adjacent meninges showed meningeal inflammation with necrosis, histiocytes, and abundant plasma cells without vasculitis; there was no evidence of neuritic plaques, neurofibrillary tangles, or amyloid angiopathy. Treatment was initiated with insertion of a ventriculoperitoneal shunt and methylprednisolone for 5 days followed by prednisone. Rapid improvement occurred, and 6 weeks later, the patient was alert, fully oriented, and talkative; the MMSE score was 29, with only sentence writing impaired. Rigidity and incontinence had resolved, and although gait remained apraxic, he could ambulate for 200 ft with a walker. He was then given mycophenolate, followed sequentially by azathioprine and methotrexate, to permit continuance of a low prednisone dose. One month after the start of treatment, CSF examination showed 2 WBCs/μL, predominantly lymphocytic, 3 RBCs/μL, protein 124 mg/dL, and glucose 44 mg/dL. After 5 months of treatment, the patient had normal cognition and gait. By 8 months, the P-ANCA titer had fallen to 1:40. A diagnosis of P-ANCA meningitis with hydrocephalus was reached for this case of reversible dementia due to a rheumatologic disease. Figure 14.1 MRI (a) T1-weighted image showing ventricular enlargement. (b) FLAIR image showing meningeal hyperintensity consistent with inflammation. SLE is a chronic, multisystem autoimmune disease with the potential to affect any organ system. Prevalence rates of SLE are up to 120 cases per 100 000 [6–8]. Incidence rates of SLE in the Americas and Europe range from 2 to 8 per 100 000/year [9, 10], with peak incidence between ages 15 and 40 years and a female to male ratio of 6: 10 to 1 [11]. Traditionally, renal and lung involvement has been thought to be responsible for the greatest morbidity and mortality in SLE [12, 13]; however, with current medical therapies, involvement in these organ systems is now associated with increasingly favorable outcomes. Instead, neuropsychiatric manifestations have emerged as a major cause of morbidity in SLE [14–17]. In 1999, the American College of Rheumatology (ACR) developed a nomenclature system for 19 neuropsychiatric syndromes in SLE (NPSLE) [18], 12 of which are considered CNS manifestations (Table 14.2). These broad criteria encompass a range of psychiatric (e.g., mood disorders) and neurologic (e.g., stroke, seizure) syndromes, and prevalence studies using this nomenclature suggest that up to 80% of patients with SLE have at least one of these NPSLE syndromes [19, 20] and the majority of patients have more than one. Table 14.2 Neuropsychiatric manifestations of SLE described by the American College of Rheumatology. Cognitive dysfunction has been observed to be the most common NPSLE manifestation in SLE, and if mild levels of cognitive impairment are included, prevalence rates reach 81% of persons with SLE. Using more conservative classification criteria of impairment, more moderate levels of cognitive impairment have been observed to affect approximately 50–66% of patients with SLE [19, 21]. Although severe cognitive impairment or dementia is relatively rare in SLE, studies have observed that this level of impairment occurs in approximately 6–8% of patients with SLE [19, 20]. Studies of SLE have observed impairment in almost any cognitive domain, but the most commonly affected include speed of information processing, attention, learning and recall, executive function, and visuospatial skills [22–29], suggesting predominant involvement of subcortical brain regions. In general, patients with SLE have been classified as having NPSLE after the onset of other NPSLE manifestations mentioned previously, and patients who have not had overt neuropsychiatric manifestations are classified as non-NPSLE. As might be expected, NPSLE patients demonstrate greater severity of cognitive impairment compared to non-NPSLE patients [23, 27, 30, 31]. However, several studies have observed that patients with non-NPSLE demonstrate more cognitive impairment than healthy matched peers [27, 29, 30, 32]. These studies suggest that in the absence of other overt neurological involvement, cognitive dysfunction may still be present in SLE, possibly heralding subtle brain involvement early in the disease course. It should be noted that some studies have not observed robust cognitive differences between non-NPSLE patients and controls [33, 34]. A number of approaches have been used to detect cognitive impairment in SLE, but the most commonly used assessment approach was developed by the ACR ad hoc committee in 1999 [18]. This expert committee assembled a 1 h neuropsychological battery that has since been validated and deemed reliable for use in SLE [21] (see Table 14.3). Table 14.3 American College of Rheumatology-recommended neuropsychological assessment battery for use in systemic lupus erythematosus. Both conventional and improved neuroimaging techniques have been used to study the associations between cognition and CNS involvement in SLE. Most of these studies have not included neuropsychological testing. Hyperintense WM lesions observed on conventional MRI are the most commonly observed neuroradiological findings in SLE, present in up to 70% of patients [35]. In general, these findings are more common among patients with antiphospholipid antibodies and manifestations of NPSLE, and WM changes have been associated with attentional dysfunction [36]. Additionally, cerebral atrophy has been observed in up to 12% of patients with SLE [37, 38]. Improved neuroimaging techniques have recently been used to examine correlations of brain abnormalities with cognitive dysfunction. One such technique that has shown promise in SLE is MR spectroscopy (MRS), with its capacity to measure WM choline (Ch) to creatine (Cre) ratios [39, 40], and diffusion tensor imaging (DTI) [41], which can examine specific WM tracts. An MRS study of frontal lobe WM in non-NPSLE, for example, demonstrated a correlation between increased Ch and measures of executive dysfunction and inattention, suggesting that immune-mediated myelinopathy may be an early pathogenic event in the cognitive dysfunction of SLE [52]. A broad range of factors have been studied in the context of cognitive decline in SLE. Autoantibody production, CV disease, inflammatory factors including cytokines, and other molecular and hormonal factors have all attracted attention as potential contributors to cognitive impairment and dementia. SLE patients have a 7.9-fold increased risk for CVD [42], and the presence of aPLs is a recognized risk factor [43–47]. APLs include a wide and heterogeneous group of immunoglobulins; however, the majority of studies in SLE have focused on LAC, aCL, and more recently the presence of β2- glycoprotein 1. The laboratory classification of aPLs has evolved, and more recently, it has been suggested that aPL positivity be considered only when there is evidence of two subsequent positive tests assessed at least 12 weeks apart [4]. In SLE, the prevalence of aCL ranges from 12 to 30% [48–50], and the prevalence of LACs from 15 to 34% [51, 52]. The presence of aPL is one factor that is consistently associated with cognitive dysfunction in SLE [53–58]. Few studies have been conducted using the more recent and more conservative laboratory classification of persistent positivity of aPLs over time or incorporating the use of β2-glycoprotein 1. In one study of a large sample of SLE patients, aPL was associated with almost a twofold risk of cognitive dysfunction; and among those patients without other overt clinical manifestations of CNS involvement, aPL was associated with a threefold risk of cognitive dysfunction [59]. The cognitive impairment resulting from aPL can be quite severe and has been observed in case series and case reports to be predictive of the onset of marked cognitive impairment and dementia [60]. The use of specific drugs for prophylaxis of CV disease among patients who are aPL positive but who lack evidence of specific neuropsychiatric events is a topic of controversy, and results of studies using aspirin for the prevention of thrombotic events in people with aPL are mixed [61–63]. Among patients with evidence of thrombosis or other vascular pathology, the key clinical management approach is typically anticoagulation, especially when there is evidence of aPL-related clinical syndromes meeting criteria for antiphospholipid antibody syndrome (discussed in the following text). Recently, there has been significant enthusiasm regarding a specific autoantibody model of cognitive impairment in SLE, specifically cognitive impairment precipitated by cross-reacting anti-NMDA NR2 (anti-NR2) antibodies. Anti-NR2 antibodies appear to be present in 20–35% of patients with SLE [64–69] and produce glutamate excitotoxicity, neural injury, and cell death in mouse models [70, 71]. Behavioral studies find memory dysfunction in these mice, and pathological studies find neuronal loss in the hippocampus [70, 71] The anti-NR2 hypothesis has been tested in five clinical studies, and the majority have found no association between these antibodies and cognition in SLE [64, 67, 69, 72]. One study did find an association between anti-NR2 antibodies and memory function [67]. These studies all employed serum autoantibody markers to detect CNS processes, but it is unclear to what extent these antibodies are transported across the blood–brain barrier. One study did investigate the presence of anti-NR2 antibodies in the CSF and found associations with diffuse neuropsychiatric syndromes in SLE [73]. Ongoing work to better understand conditions that promote blood–brain barrier permeability in SLE is underway and may improve the understanding of the transport of anti-NR2 antibodies into the CNS. Such information would directly inform the use of therapies targeted at the NMDA receptor. Other autoantibodies have long been studied as pathogenic mechanisms for cognitive impairment in SLE. The role of antiribosomal P antibodies has been a target of investigation, but no clear associations have been found between this autoantibody and cognition in SLE [74]. Inflammatory markers have also been under investigation as factors precipitating cognitive dysfunction, and one study observed an association between serum IL-6 [75] and C-reactive proteins [76]. Additionally, neuropeptide Y has also been implicated in clinical studies of cognition in SLE [77]. It is well known that SLE is characterized by accelerated cardiovascular and CV disease. This increased burden may be partially explained by the increased prevalence of traditional CV risk factors compared to non-SLE peers (e.g., tobacco use, hypertension, hyperlipidemia, obesity, and sedentary lifestyle) [78–81]. However, many studies also cite comparable rates of traditional CV risk factors compared to peers [79, 82]. Traditional risk factors do not fully explain the CV burden in SLE [42], and disease- and treatment-related factors must also be considered [83–85] (e.g., inflammation, chronic glucocorticoid use, etc). SLE is often considered to be the prototypical autoimmune disease, and its neuropsychiatric manifestations are likely multifactorial in etiology. Although SLE is the rheumatologic condition whose cognitive and neuropsychiatric manifestations have received the most attention, the etiology of cognitive decline remains unclear. Through the ongoing study of large and diverse cohorts of SLE patients, a more standardized approach to the detection of cognitive decline, and advances in neuroimaging, it is likely that our understanding of these syndromes will be significantly improved. The term antiphospholipid syndrome (APS) was coined to describe the clinical combination of the presence of aPL and a specific syndrome of arterial or venous thrombosis [86]. When APS occurs in the absence of SLE or other connective tissue disease, it is called primary APS. Primary APS occurs in 0.5–6% of the general population [87, 88]. Secondary APS is diagnosed when a patient has a diagnosis of another condition (most often SLE) and has aPL with evidence of hypercoagulability. Approximately 30% of patients with SLE have concomitant APS [87]. The average age at APS diagnosis is 34 with only 12% of patients diagnosed after age 50. The male to female ratio for primary APS is 1:3.5, and for secondary APS, it is 1:7 (more closely approximating the gender ratio for SLE) [49]. The primary clinical syndromes that can occur as a result of APS include thrombosis and pregnancy morbidity. Neurological manifestations of APS lead to the greatest morbidity and mortality. The most common and severe manifestation of APS is stroke, occurring in approximately 19% of patients, followed by transient ischemic attack in 11% [89, 90]. Other reported CNS sequelae have included migraine, cognitive dysfunction, seizures, chorea, transverse myelitis, psychosis, depression, and Guillain–Barré syndrome [91]. Rarely, patients can develop catastrophic APS, characterized by rapid development of multiple microthrombi in multiple organ systems (brain, kidney, lung, skin); mortality in catastrophic APS nears 50% [92]. Dementia is seen in less than 5% of patients with APS, but mild to moderate levels of cognitive dysfunction occur in 33–43%; these patients generally present with impairments in memory and learning, language, attention, and executive functions [93, 94]. In a study of 30 patients with APS-associated dementia, 47% of patients had primary APS, 30% had SLE, and 23% had characteristics of SLE [60]. Thirty-seven percent of this cohort had suffered a stroke, and 33% had Sneddon’s syndrome (described in the following text). Notably, 37% did not present with other APS manifestations before to the diagnosis of dementia, suggesting that laboratory studies of aPL may be warranted among young individuals who present with unexplained cognitive impairment. MRI findings in APS include multiple foci of WM hyperintensities, and occasionally, confluent areas of hyperintensity are observed [95]. MRS has shown a decreased ratio of N-acetylaspartate (NAA) to Cre and an increased Ch to Cre ratio [95]. The degree to which the MRS metabolic studies predict APS beyond MRI hyperintensities is unclear, but the use of NAA/ Cre ratios in SLE patients with APS may be a promising clinical method to quantify the course of neurological and neurobehavioral involvement [96]. Sneddon’s syndrome is a related condition characterized by the development of CV disease (stroke and transient ischemic attacks) and concomitant livedo reticularis. The onset of CV disease occurs at a relatively young age, generally before age 45 [97]. Over 40% of those with Sneddon’s syndrome have APS [98]. Neuroradiological findings suggest that Sneddon’s syndrome presents with leukoaraiosis and lacunar infarcts [99] more frequently than primary APS, which is more typically characterized by larger arterial vascular lesions. The clinical course of Sneddon’s syndrome is generally worse than APS, particularly with the recurrence of CV events. Greater than two-thirds of patients have cognitive impairment [100, 101], and cognition appears to deteriorate as CV events accumulate [102, 103]. Progressive cognitive decline and dementia in the absence of discrete events have also been reported in younger adults with Sneddon’s syndrome [104, 105]. Clinical management of the neurologic aspects of Sneddon’s syndrome is somewhat controversial, but anticoagulation remains the most commonly used treatment followed by other immunosuppressive drugs including glucocorticoids and cyclophosphamide [106].
Rheumatologic and other autoimmune dementias
Introduction
The diagnostic conundrum of rheumatologic disease in the neurologic setting
Condition
Potential neurological manifestations
Primary extraneurological manifestations
Potential behavioral characteristics
Systemic lupus erythematosus
See Table 14.2; 19 identified peripheral and central nervous system manifestations (e.g., seizure, stroke, headache)
Any organ system (commonly dermatologic, renal, and musculoskeletal involvement)
Cognitive impairment
Confusion/disorientation
Depression and/or anxiety
Psychosis
Antiphospholipid syndrome
Stroke
Pregnancy morbidity
Cognitive impairment
Transient ischemic attack
Evidence of vascular thrombosis in any tissue or organ
Psychosis
Migraine
Depression
Seizures
Chorea
Transverse myelitis
Guillain–Barré syndrome
Sneddon’s syndrome
Stroke
Livedo reticularis
Cognitive impairment
Transient ischemic attack
Sjögren’s syndrome
Transverse myelitis
Dryness of the eyes, mouth, and other mucous membranes
Cognitive impairment
Meningoencephalitis
Subarachnoid hemorrhage
Migraines
Antineutrophil cytoplasmic antibody-associated vasculitides (e.g., Wegner’s granulomatosis, Churg–Strauss syndrome)
Hemorrhagic stroke (especially in the context of uncontrolled hypertension)
Wegner’s granulomatosis
Cognitive impairment
Peripheral nerve involvement
Upper respiratory involvement (including inflammatory lesions in sinus regions)
Disorientation
Other pulmonary involvement
Renal involvement
Churg–Strauss Syndrome
Upper and lower respiratory
Cardiac complications
Behcet’s disease
Headache
Psychosis
Dysarthria
Confusion
Cranial nerve findings
Cognitive impairment
Ataxia
Giant cell arteritis
Headache (especially in temporal regions)
Ophthalmologic signs
Confusion
Cranial nerve involvement
Polymyalgia
Cognitive impairment
Neuro-ophthalmologic signs
Systemic symptoms (weight loss, fatigue, etc.)
Neuropathy
Medium vessel
Stroke
Scalp tenderness
Transient ischemic attack
Temporal artery tenderness
Jaw claudication
Large vessel
Claudication of arms, asymmetrical pulses
Systemic sclerosis (scleroderma)
Cranial and peripheral neuropathy
Sclerotic skin
Cognitive impairment
Rarely cerebrovascular involvement (especially in the context of renal, heart, and pulmonary involvement)
Reynaud’s phenomenon
Sarcoidosis
Cranial neuropathy
Lung, skin, and eye manifestations
Cognitive impairment
Meningeal involvement
Neuroendocrine involvement (in response to hypothalamic and pituitary involvement)
Psychosis
Seizures
Delirium
Celiac disease
Ataxia
Chronic diarrhea
Cognitive dysfunction
Headaches
Fatigue
Confusion
Encephalopathy
Seizures
Chorea
Case report
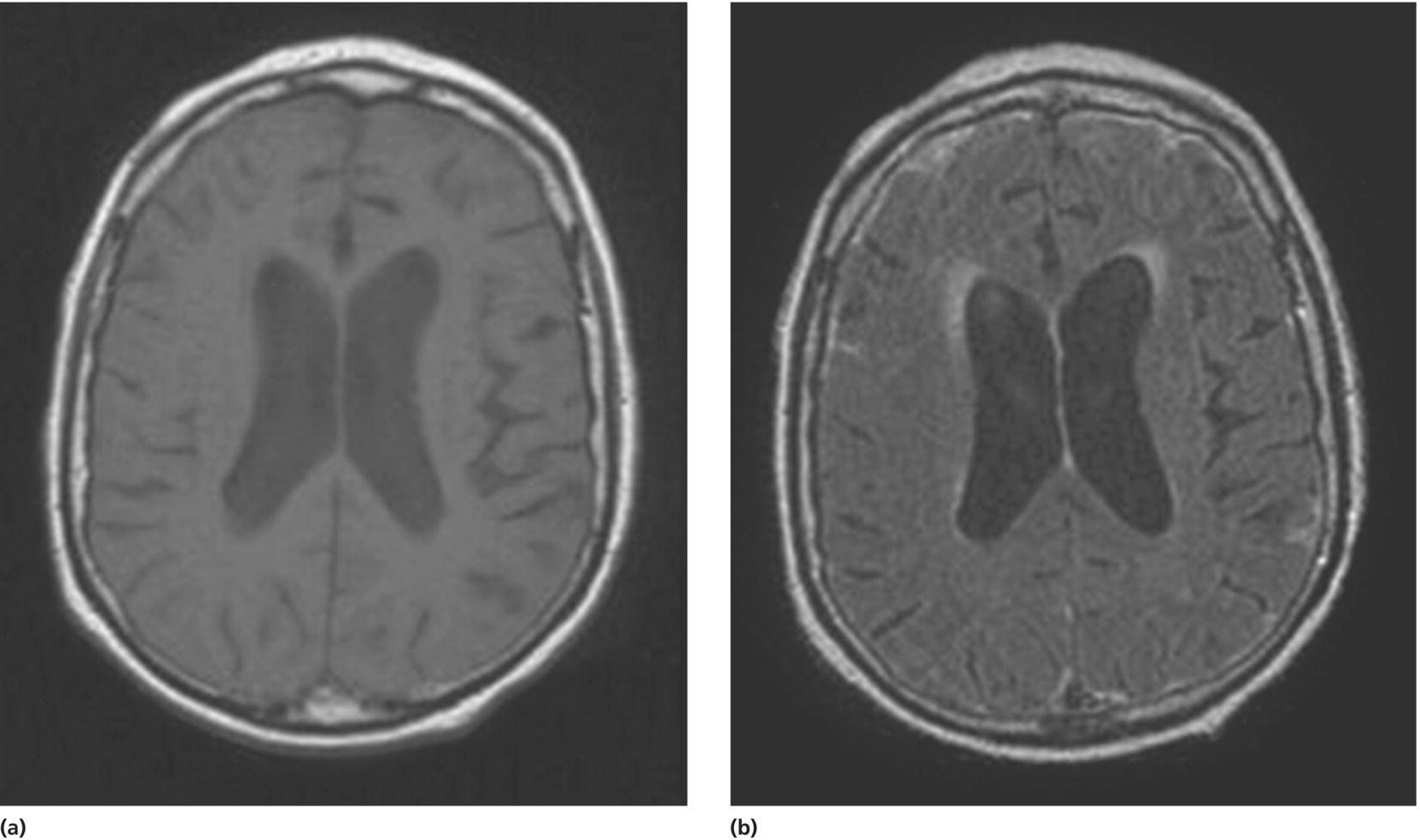
SLE
Central nervous system manifestations
Peripheral nervous system manifestations
Seizure disorders
Autonomic disorder
Cerebrovascular disease
Myasthenia gravis
Demyelinating syndrome
Neuropathy, cranial
Aseptic meningitis
Mononeuropathy, single or multiplex
Headache
Acute inflammatory demyelinating polyradiculoneuropathy
Movement disorder (chorea)
Plexopathy
Myelopathy
Polyneuropathy
Acute confusional state
Anxiety disorder
Cognitive dysfunction
Mood disorder
Psychosis
The nature of cognitive impairment in SLE
Cognitive domain
Measure
References
Executive function
Response inhibition
Stroop Color–Word Interference Test
[177–180]
Verbal fluency
Phonemic Fluency Test
[181]
Sequencing/shifting
Trail Making Test Part B
[182]
Working memory
Letter–Number Sequencing Test
[183]
Processing speed
Digit Symbol Substitution Test
[183]
Verbal learning and recall
California Verbal Learning Test II
[184]
Nonverbal learning and recall
Rey–Osterrieth Complex Figure Test
[185, 186]
Semantic fluency
Animal Naming Test
[187]
“Premorbid” verbal ability
North American Adult Reading Test
[188]
Fine motor speed and manipulation
Finger tapping test
[189]
MRI findings in SLE
Mechanisms of cognitive decline in SLE
Antiphospholipid antibodies (aPL)
Cross-reacting N-methyl-D-aspartate (NMDA) receptor antibodies
Other mechanisms under consideration
Summary
Antiphospholipid syndrome and Sneddon’s syndrome
Sjögren’s syndrome
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


