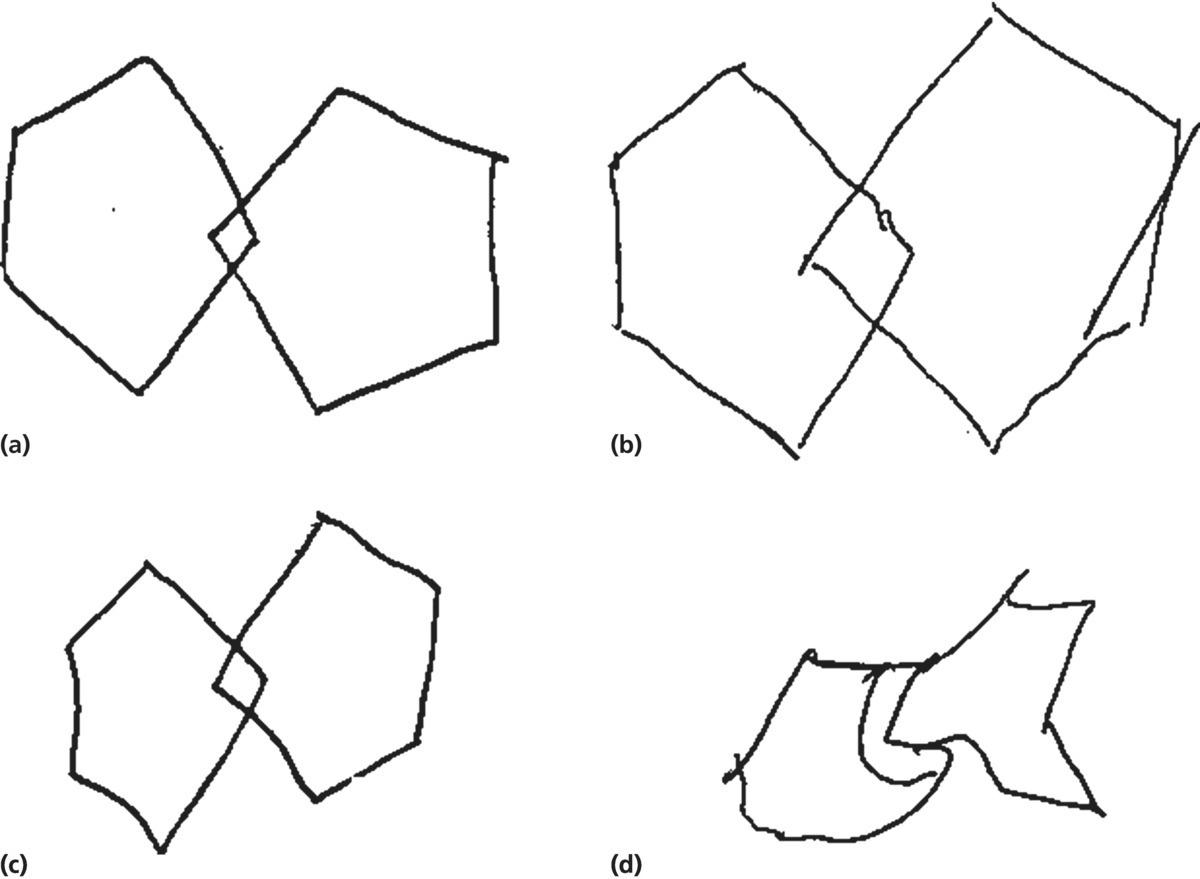
CHAPTER 6 Carol F. Lippa1 and Katherine L. Possin2 1 Drexel University College of Medicine, Philadelphia, PA, USA 2 University of California, San Francisco, San Francisco, CA, USA Dementia is characterized by cognitive impairment severe enough to interfere with daily functioning [1]. Alzheimer’s disease (AD) is the most common dementia subtype, accounting for more than half of all dementia cases. In AD, memory loss is the most significant aspect of cognitive impairment [2]. Dementia with Lewy bodies (DLB) is probably the second most common dementia, affecting nearly one-quarter of dementia subjects. In contrast to AD, core diagnostic criteria of DLB include visual hallucinations, parkinsonism, and fluctuations of attention and alertness (see Table 6.1). These symptoms are rare in cognitively normal elderly individuals and early AD patients [4]. Other features in DLB include rapid eye movement (REM) sleep behavior disorder (RBD), other sleep dysfunctions, syncope, transient impairment in consciousness, delusions, depression, and early incontinence [5–7]. The diagnosis of DLB is important to make because treatment specifics differ from those used in other dementias. For example, DLB patients are more susceptible to neuroleptic hypersensitivity than AD patients [8]. Table 6.1 Comparison of DLB and PDD. Adapted from McKeith [3] and Emre [4]. Central feature Core features (two sufficient for probable, one for possible) Suggestive features (one or more + in presence of one or more core features sufficient for probable; one or more but without any core features sufficient for possible) Supportive features (commonly present, but no diagnostic specificity) Less likely if Core features (both must be present for probable diagnosis) Associated clinical features Less likely if (don’t exclude but diagnosis uncertain) Features suggesting other conditions (impossible to reliably diagnose PDD) Patients with Parkinson’s disease (PD) often develop cognitive decline and up to 80% will progress to dementia (Parkinson’s disease with dementia (PDD), [4]). PDD differs from DLB in the temporal sequence of initial symptoms, but with progression, the syndromes and pathology become similar. Both involve progressive cognitive decline involving visuospatial, attentional, and executive functions. Psychiatric disturbances include visual hallucinations, anxiety, apathy, and depression. The motor symptoms are similar in PDD and DLB, and both have more axial symptoms, postural instability, and gait difficulty than nondemented PD [9]. Both are characterized pathologically by intracytoplasmic inclusions containing ubiquitin and alpha-synuclein called Lewy bodies (LBs). PDD and DLB brains have a loss of the neurotransmitters acetylcholine and dopamine [10]. PD was first profiled in 1817 by James Parkinson in “An Essay on the Shaking Palsy.” He described the classic motor symptoms of PD but reported “the senses and intellects being uninjured” [11]. It is now increasingly recognized, however, that many patients newly diagnosed with PD have cognitive impairment associated with their disease and that many of these patients will go on to develop a dementia syndrome. The initial autopsy descriptions of patients with DLB were written by Dr. Okazaki [12]. They described patients with progressive dementia but without the motor symptoms of PD whose brains had cortical LBs. This was followed by detailed clinicopathologic descriptions of DLB cases by Kenji Kosaka [13] who reviewed all available literature cases and noted a characteristic clinical syndrome. Since the original descriptions, immunohistochemistry with antibodies to components of LBs (antiubiquitin and anti-alpha-synuclein antibodies) has made diagnosis easier, and we now realize that DLB is a common disorder. In 1996, McKeith et al. established the first consensus criteria for DLB and formalized the term “dementia with Lewy bodies.” The discoveries of α-synuclein mutations in families with autosomal dominant PD [14] and of α-synuclein as the main component of LBs [15] linked PDD and DLB from a biologic standpoint. The DLB/PDD Working Group recommended the use of a single term and model “Lewy body disorders” (LBD), encompassing these two syndromes and nondemented PD, to study disease pathogenesis, new treatments, and biomarkers [16]. Multiple system atrophy (MSA) is a rare neurodegenerative disorder that, as with DLB and PDD, is related to a disturbance of alpha-synuclein, although, in MSA, inclusions are within glial cells, rather than neurons. An in-depth description of MSA is outside the scope of this chapter; however, the syndrome is characterized by a combination of autonomic, parkinsonian, and cerebellar features. When parkinsonian features predominate, the term MSA-P is used; when cerebellar features predominate, the term MSA-C is used. Dementia is infrequent and in fact is listed as a nonsupportive feature in current consensus criteria [17]. Less severe cognitive impairment is common in MSA, however, and might be intermediate in severity to that of DLB and PD patients of similar disease duration [18]. Approximately 1% of the population over 60 years old suffers from PD, increasing to 4% in older age groups, with slightly higher prevalence in men than women [19, 20]. Approximately 30% of PD patients are estimated to have dementia, which is four to six times higher than controls [21]. Longitudinal PD cohort studies suggest up to 80% of PD patients exhibit dementia before death [22]. About 15–20% of dementia cases involve DLB, whereas approximately 6% are due to PDD [23]. Monogenetic forms of LB disorders represent less than 10% of LBD cases; the majority of cases result from interactions among susceptibility genes and environmental risk factors [24]. A recent case–control study looking at 19 risk factors in 147 DLB subjects compared with cognitively normal controls, as well as an AD cohort, found that compared to controls, DLB subjects were more liked to have depression, history of anxiety, and a family history of PD and to carry ApoE4 alleles, but were less likely to have cancer or use caffeine. Compared to AD subjects, DLB subjects were more likely to be male, about 2 years younger (72.5 vs. 74.9), highly educated, have a family history of PD, have no ApoE4 alleles, and have had an oophorectomy before age 45 [25]. A 74-year-old gentleman without significant past medical history was referred for neurological consultation for a 2-year history of difficulties concentrating. He lost track of conversations, had trouble reading, and had problems getting lost. His general intellect was intact; he reported no memory loss and had no difficulties with specific activities but felt he was less productive. Irritability was his only behavioral symptom. He was on no medications. Family history was notable only for a brother with PD who had a L-DOPA-responsive tremor. General physical examination was normal. Neurological examination disclosed an alert, pleasant patient. His Mini-Mental State Examination [26] (MMSE) score was 25/30, losing 1 point for recall, 3 points for spelling WORLD backward, and 1 point for copying the pentagons. Language skills and comprehension were intact, but he had problems with attentional tasks. Cranial nerve, motor, and gait examination was normal, with the exception of an abnormal glabellar response. Head CT and blood work for reversible causes of dementia was normal. An atypical presentation of early stage of AD was suspected, and an annual follow-up was recommended. One year later, he had decreased activities of daily living, sometimes requiring help in cutting his food, needing his clothes laid out for him, occasionally putting on a garment backward or inside out, and often shaving incompletely. He was occasionally visually disoriented at home and had more difficulty with routine tasks such as shoveling snow and piling it in illogical places. Once, he walked the dog but returned home without him. He slept more and took daytime naps. Behaviorally, he was mildly depressed, was quieter, and was having well-formed, usually visual and occasionally threatening hallucinations typically involving people from his past. He imagined that his (deceased) brother was living with them and insisted on making plans for him to have a bedroom. He stumbled and his gait was slower. He was alert but looked “a little lost.” Vital signs, including orthostatics, and general physical examination remained normal. Neurologically, he was pleasant and cooperative but hypophonic. His MMSE was 15/30, losing 5 points on orientation—1 for registration, 1 for delayed recall, 5 for spelling the WORLD backward, 1 for a 3-step command, 1 for copying the pentagons, and 1 for repetition. Most language skills and comprehension were intact. Cranial nerve examination was remarkable only for a mild restriction in upgaze. Motor examination revealed normal strength, bulk but increased muscle tone with mild cogwheel rigidity, and symmetrically brisk reflexes. There was no resting tremor, but a slight postural tremor. Gait testing revealed a mildly stooped posture, shuffling, and reduced stride length. Plantar responses were flexor, but marked frontal release signs developed including a suck, snout, and palmomental responses as well as bilateral grasp reflexes. Sensory examination was intact to primary and cortical sensory modalities. He was diagnosed with an extrapyramidal dementia of unclear etiology. He progressed gradually and died 2 years later, 5 years after onset. His brain weight was normal (1330 grams) and showed no focal atrophy with normal ventricle size. The substantia nigra and locus ceruleus were depigmented. LBs were frequent including the amygdala and the adjacent entorhinal cortex/parahippocampal gyrus and were also present in the cingulate gyrus and lower layers of the frontal, temporal, and parietal lobe cortex. The substantia nigra and locus ceruleus showed neuronal loss and LBs in some remaining neurons. There were no neurofibrillary tangles (NFTs), but numerous diffuse amyloid plaques were scattered in the cerebral cortex, and moderate amyloid angiopathy was seen. Although there was some Alzheimer’s pathology, as he did not have NFTs, he did not meet NIA-Reagan or CERAD criteria for AD [27]. His final pathological diagnosis was DLB. Diagnostic criteria specific for dementia associated with PD (PDD) were proposed in 2007 by the Movement Disorder Society Task Force [4]. Diagnosis of PDD requires a dementia syndrome with insidious onset and slow progression, developing with the context of established PD (Table 6.1). The dementia must involve a decline in multiple cognitive domains severe enough to impair daily life. A probable diagnosis requires impairment in at least two of the following: attention, executive functions, visuospatial functions, or memory retrieval. Behavioral features including apathy, changes in personality and mood including depression or anxiety, hallucinations, delusions, or excessive daytime sleepiness support the diagnosis. Diagnostic criteria for DLB were proposed in 1996 [28] and refined in 2005 [3] by the DLB Consortium (Table 6.2). According to the original criteria, probable DLB is characterized by dementia associated with any two of the following three core features: (i) fluctuating cognition or level of consciousness, (ii) visual hallucinations, and (iii) spontaneous parkinsonian motor signs. The refined criteria include suggestive features: RBD, neuroleptic sensitivity, and low dopamine transporter uptake in the basal ganglia demonstrated by SPECT or PET imaging. Probable DLB can also be diagnosed if one or more of these suggestive features are present along with one or more core features. The new recommendations contain provisions for a probabilistic basis for the pathologic diagnosis of DLB based on the predominance of cortical and limbic LBs relative to the density of neurofibrillary tangles. Cases with LBs in the setting of extensive AD-type pathology are classified as having a “low likelihood” of DLB. Table 6.2 Bedside tests to evaluate cognitive features in PDD/DLB. Motor signs in DLB and PDD include the classic triad of akinesia, rigidity, and tremor, which might be responsive to dopamine replacement, and axial symptoms, which are considered less responsive [4]. Extrapyramidal motor symptoms are present in about half of DLB patients at presentation, and they eventually occur in most of the remaining patients [29]. In comparison to PD, in DLB, there usually is more axial rigidity and postural instability. Gait is disrupted earlier in DLB than PD, and falls are not uncommon. Although resting tremor is a common presenting symptom in PD, DLB patients more typically have an intention or position tremor, if present at all. Their rigidity might lack the classical cogwheel quality that is the hallmark of PD. The gait of DLB patients is similar to PD/PDD and includes postural instability, a stooped posture, and festination [9]. Deep tendon reflexes are generally symmetrical in DLB, and frontal release signs might include palmomental responses and a glabellar response. In PDD, patients often present with asymmetrical resting tremor and other classical features of PD including cogwheel rigidity. Postural instability and gait disorder is associated with accelerated cognitive decline and subsequent dementia, whereas patients with tremor less commonly develop early dementia [30]. Clinical assessment of motor dysfunction is difficult when dementia is severe; however, a subscale of the Unified PD Rating Scale [31] contains five items (resting tremor, action tremor, bradykinesia, loss of facial expression, and rigidity) that can be reliably assessed independent of dementia severity. The neuropsychological phenotype of both DLB and PDD is characterized by impairments in visuospatial and executive functions and also by fluctuations in attention and arousal, with core language functions relatively preserved [32–34]. When matched for dementia severity, the cognitive profiles of PDD and DLB are similar or indistinguishable [35, 36], although some studies suggest greater attentional impairment in DLB [37]. In comparison to AD, LB dementia patients show worse visuospatial, attentional, and executive impairments, whereas AD patients show more severe memory impairment [38]. An absence of visuospatial impairment early in the course of dementia is unusual in LBD and suggests a different etiology [39]. Figure 6.1 presents an example of figure copy performance in DLB that illustrates visuospatial impairment. Figure 6.1 Patients were asked to copy the image exactly. Image (a) is the original figure that patients were asked to copy. Image (b) is the reproduced image from an 80-year-old patient with Alzheimer’s disease. Note that the patient struggles with reproducing one of the pentagons but the spatial aspects of the figure are easily identifiable. Image (c) is from a cognitively intact 82-year-old elderly individual. Image (d) shows an attempt by an 80-year-old patient with probable DLB; although cognitive symptoms were mild, he is unable to reproduce the spatial aspects of the figures. PD patients do not evidence dementia at onset; there is, however, increasing recognition that milder cognitive impairment is common [40]. Working memory, selective attention, inhibitory processing, cognitive flexibility, and learning are often impacted early and attributed primarily to nigrostriatal dopamine loss causing disruption of frontal-striatal circuitry function [41], although depletion in noradrenergic and cholinergic neurotransmitter systems have also been implicated [42, 43]. The emergence of impairment on tasks with a more posterior cortical basis (figure copy or semantic fluency) might signal the transition of the disease from the brainstem to the neocortex and indicate that progression to dementia is likely [44]. Bedside cognitive testing is useful to gauge the severity of cognitive impairment and its response to cholinesterase inhibitors, to diagnose cognitive impairment in PD, and to differentiate DLB from other diseases. The MMSE [26] is not sensitive to LBD because it emphasizes language and memory over executive function and visuospatial skills [45]. The Montreal Cognitive Assessment (MoCA; www.mocatest.org) is more sensitive to LBD; the recommended cutoff for dementia in PD is 20/30 [46]. Suggested methods for brief assessment are listed in Table 6.2. Patients exhibiting cognitive impairment on a brief exam might require more detailed neurocognitive testing. Neuropsychiatric disturbances are common in LBD and contribute to reduced quality of life [47], caregiver distress [48], and increased risk for nursing home admission [49]. In a study of 537 patients with PDD using the Neuropsychiatric Inventory (NPI), the most common symptoms were depression (58%), apathy (54%), anxiety (49%), and hallucinations (44%). Depression is more common in LB dementias than in AD [50] and equally common in PD, PDD, and DLB [5]. Depression is listed as a supportive feature in DLB criteria [3]. Anxiety often predates PD by many years [51]. Panic disorder, generalized anxiety disorder, and social phobia are prevalent, and depression is often comorbid [52]. Apathy is common in PD and DLB and is associated with cognitive decline [53, 54]. The pathophysiology of depression, anxiety, and apathy in LB dementia might involve reductions in dopamine, noradrenaline, and serotonin [55–58], and depression has been associated with LBs in the amygdala [59]. Although depression, anxiety, and apathy are frequent in other dementias, visual hallucinations are more specific to LBD [60]. Hallucinations occur in 45–65% of PD and 60–80% of DLB patients, with much lower rates in early AD (4–8%) [4]. In patients with PDD or DLB and hallucinations, most experienced daily complex hallucinations (i.e., people or animals) [61]. Auditory and tactile hallucinations less commonly occur. Hallucinations in DLB are associated with LBs in the temporal cortex [62] and with hypoperfusion on single-photon emission tomography in the left ventral occipital gyrus and bilateral parietal areas [63]. Hallucinations are associated with cholinergic deficits [64] and often respond to cholinesterase inhibitors. Delusions are also common in LB dementia. Capgras syndrome, for example, is characterized by the delusional belief that a person, usually someone who is closely related, has been replaced by an imposter. For example, a patient might say “she looks like my wife, but she is not my wife.” When occurring early, Capgras syndrome is suggestive of LB dementia; however, it also occurs in late-stage AD. In a study of 47 patients with Capgras syndrome, 38 were clinically diagnosed with a neurodegenerative disease, and of those, 26 had LBD, whereas only 7 had AD (only two subjects were pathology proven, however) [65]. Recognition of premotor and predementia phases of LBD, which might span 20 years or more, is guiding the search for predictive biomarkers as well as risk factors. RBD is a common early feature of synucleinopathies and has been estimated to precede PD and DLB by a decade on average [66–68], with some patients showing clear RBD up to 50 years before LBD has clinically manifested [69]. Infrequent bowel movements were associated with the development of PD, with a mean 10-year latency [70], and in a large aging cohort without PD or dementia, infrequent bowel movements were associated with a fourfold risk of incidental LBs at autopsy [71]. Psychiatric symptoms (especially anxiety) might also predate PD [51, 72, 73]. High scores on the composite neuroticism scale of the Minnesota Multiphasic Personality Inventory also predict PD even when administered at ages 20–39 years [73].
Lewy body dementias (DLB/PDD)
Introduction
DLB
PDD
Nosology
Historical issues/nomenclature
Epidemiology
Case study
DLB/PDD clinical features
Diagnostic criteria
Attention: serial 7 s, months, or days backward
Memory: learning a word list with delayed free recall, recall versus recognition
Executive functions: verbal fluency, trail making, clock drawing
Visuospatial functions: copying intersecting pentagons or a three-dimensional cube
Language: confrontation naming, understanding complex sentences
Neurological exam and motor features
Cognitive features
Neuropsychiatric features
Preclinical symptoms
Comorbidities
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


