CHAPTER 8 Praveen Dayalu1, Roger L. Albin1,2 and Henry Paulson1 1 University of Michigan, Ann Arbor, MI, USA 2 VAAAHS GRECC, Ann Arbor, MI, USA More than 20 neurological disorders are caused by DNA repeat expansions, many of which are associated with neurodegeneration. Relatively few of these disorders, however, will be considered in the patient undergoing evaluation for atypical dementia. The principal reason why these diseases are not high on the differential diagnosis of dementia is that most heritable repeat expansion disorders do not include prominent cortical or subcortical loss as a primary site of degeneration. The most notable exception to this is Huntington’s disease (HD), in which progressive cognitive impairment is a hallmark feature. HD is the most common among the nine diseases known to be caused by polyglutamine-encoding CAG repeat expansions. The other eight polyglutamine diseases include six dominantly inherited spinocerebellar ataxias (SCAs 1,2,3,6, 7, and 17), dentatorubral-pallidoluysian atrophy (DRPLA), and the motor neuron disorder, spinobulbar muscular atrophy (SMA). Of the non-HD polyglutamine disorders, only SCA17 typically manifests with progressive cognitive impairment that culminates in dementia, although about a third of SCA2 patients develop dementia as well [1–4]. Whereas most patients with SCA17 also have significant ataxia and cerebellar atrophy, the cognitive symptoms often begin early and sometimes dominate the clinical picture before cerebellar or basal ganglionic signs surface. Other SCAs including SCA1, SCA2, SCA3, and SCA8 can also have cognitive deficits, albeit milder than SCA17, but will not be discussed in this chapter. The other relatively common repeat expansion disease associated with age-related dementia is fragile X-associated tremor/ataxia syndrome (FXTAS), which is caused by a modest noncoding CGG expansion. We begin with a detailed overview of HD because it is an important and readily diagnosed form of atypical dementia associated with basal ganglionic signs and symptoms. In discussing the differential diagnosis of HD, we touch on other disorders involving the basal ganglia in which cognitive impairment can be prominent, including the much rarer Huntington’s disease-like 2 (HDL2) disorder, which is caused by a repeat expansion in a different gene (junctophilin-3). We then briefly discuss SCA17 as the primary SCA in which dementia is a very common, and sometimes presenting, feature. Finally, we discuss FXTAS, which displays broad heterogeneity in phenotype, with progressive movement disorder, ataxia, and cognitive impairment being the most common manifestations. When discussing FXTAS, we remind the reader that among sporadic forms of progressive ataxia associated with cognitive impairment, multiple system atrophy (MSA) is perhaps the most common disorder. In 1872, physician George Huntington reported a familial chorea on Long Island, noted previously by his father and grandfather, also physicians [5]. More than a century later, Huntington’s vivid writings are a remarkably complete description of the disease that now bears his name. He described chorea as “dancing propensities” in which there “seems to exist some hidden power, something that is playing tricks, as it were, upon the will.” At first, this inherited disease began “as an ordinary chorea might begin, by the irregular and spasmodic action of certain muscles as of the face, arms, etc. These movements gradually increase when muscles hitherto unaffected take on the spasmodic action….” He noted that the disease was “confined to … a few families … hardly ever manifesting itself until adult or middle life, and then coming on gradually but surely, increasing by degrees, and often occupying years in its development, until the hapless sufferer is but a quivering wreck of his former self.” Huntington also commented on the mind, which “becomes more or less impaired, in many amounting to insanity while in others, mind and body both gradually fail until death relieves them of their sufferings.” Regarding the inheritance pattern, he noted: “When either or both parents have shown manifestations … one or more of the offspring almost invariably suffer from the disease … but if by any chance these children go through a life without it, the thread is broken….” Finally, he noted the relentless, fatal course: “I have never known a recovery … it seems at least to be one of the incurables.” HD is the most common polyglutamine neurodegenerative disease. In the United States and Canada, approximately 30 000 people carry the diagnosis, and an estimated 150 000 more are at risk. HD is most prevalent in those of European descent, approximately 10–15/100 000 [6, 7]. Pockets of particularly high prevalence, probably due to founder effects, include the Lake Maracaibo region of Venezuela and regions of Scotland and Tasmania [8–10]. Prevalence rates are much lower in populations of non-European ancestry [11]. Males and females are equally at risk. Median age of diagnosis approximates 40, with a wide range in age of onset. Onset before age 20 or after age 65 is relatively rare. Death generally occurs 15–20 years after diagnosis [12]. The combination of typical midlife onset and dominant inheritance pulls entire families down the social scale and devastates the lives of patients, mutation carriers, and unaffected family members alike. HD is an autosomal dominant disorder. In the general population, there are on average 17–20 CAG repeats in the HTT gene [13]. With a CAG repeat of 40 or more, a person will develop HD with 100% certainty, but with repeats of 36–39, there is incomplete penetrance. CAG repeat lengths of 6–26 do not cause disease and are thus considered “normal.” The intermediate range, from 27 to 35 repeats, does not cause HD with a few reported exceptions [14]. Notably, all alleles of 27 repeats and higher are unstable and prone to expand in future generations, particularly when transmitted by a male parent. Whereas the great majority of HD patients have an affected parent, up to 10% of cases result from new expansions into the disease range [15, 16]. In European populations, there are specific HTT haplotypes, involving polymorphisms in other regions of the gene, that might promote CAG repeat instability [17]. The appearance of earlier and more severe symptoms in successive generations, known as “anticipation,” reflects CAG instability with further expansions from one generation to the next. Sonia H, a waitress aged 42, started drinking whiskey every evening. At first, her husband and sons thought this was related to arguments with her coworkers. A year later, her family noted that she often “flew off the handle” at minor issues. Her boss noted a new tendency to drop platters and break dishes. Others suspected her of illegal substance abuse because of her mildly slurred speech and increasing fidgetiness. At age 44, after she struggled to plan her traditional Thanksgiving dinner, her family brought her to a neurologist. Sonia’s parents were alive and healthy in their 70s. Her older brother was also well. She knew of no more distant family members with a movement disorder, dementia, or prominent psychiatric problems. Her only medications were citalopram for “moods” and omeprazole. She did not smoke. Other than intermittently heavy alcohol, she denied substance abuse. In the office, she was irritable toward her family, offering curt denials of their reports. When her son described her new compulsion to count cash at home, she snapped, “It isn’t your money, so what do you care!” Her speech was irregular in volume and emphasis. She denied involuntary movements, but her family thought she looked “restless.” Throughout the interview, there were occasional brief random movements: pursing of her lips, raising of her eyebrows, tilting of her head, extending or flexing a finger or wrist, and inverting a foot. On the Montreal Cognitive Assessment (MoCA), she scored 23/30. She lost 1 point each for trails test, serial 7s, digits backward, delayed recall, and sentence repetition. She lost 2 points for concrete responses on abstraction. She could not complete the Luria maneuver unless cued verbally: “fist, edge, palm.” On motor examination, her involuntary movements were brought out when she counted backward with hands outstretched; in particular, there were “piano-playing” finger movements. She blinked and moved her head when initiating saccades. She could not keep her tongue protruded for a full 10 s; it flicked briefly back into her mouth at 8 s. Finger tapping was slightly slowed on the left. Hand pronation–supination was clumsy bilaterally. Heel–knee–shin was slightly irregular bilaterally. Her gait was stable and narrow based but with occasional random shoulder, trunk, and hand movements creating a vaguely puppetlike appearance. She sidestepped twice in a 10-step tandem walk. On pullback testing, she took 3 steps to maintain her balance. The neurologist concluded that Sonia had chorea, mild ataxia, and cognitive deficits typical of executive dysfunction. Given her change in personality and behavior, he was most concerned about HD. The lack of a family history, however, was puzzling. His differential diagnosis also included other heredodegenerative diseases (Wilson’s disease, neuroacanthocytosis, DRPLA, SCA17), inflammatory disorders (SLE, antiphospholipid antibody syndrome, demyelinating disease, CNS angiitis), unrecognized substance abuse with complications, thyroid disease, or chronic CNS infection (e.g., HIV). Vitamin B12 level, TSH, and a comprehensive metabolic panel had been unremarkable. The neurologist advised the patient and her family that HD was the most common cause of adult-onset chronic chorea. He informed them that if HD testing was clearly negative, he would initiate a broader workup including brain MRI. The patient and her family were referred to a genetic counselor to discuss the implications of testing for the HD gene. Testing revealed 44 CAG repeats in one HTT allele and 17 in the other. Sonia received the diagnosis of HD at a subsequent visit, with her family present. Her teenage sons were informed that they, too, were at risk for HD, though they were asymptomatic. The neurologist informed them that after reaching age 18, they could receive predictive testing if they so chose. As to why both of Sonia’s elderly parents were unaffected, the neurologist explained that one of them might carry an unstable CAG repeat expansion of less than 40 that was not fully penetrant. Other explanations could include nonpaternity. Sonia’s citalopram was increased to 40 mg per day to help with irritability, anxiety, and obsessions. She was referred to occupational therapy to help her compensate for her decline in coordination. Her husband asked for treatment of her “restlessness”; however, the neurologist noted that Sonia herself was not aware of or disabled by her mild chorea, so he persuaded the family that antichorea therapy was not necessary at this time. He informed the family of support and advocacy resources in HD, as well as research opportunities at the nearby academic medical center. The classic clinical triad in HD is (i) a progressive motor disorder, notably with chorea but also with varying degrees of dystonia, bradykinesia, rigidity, ataxia, dysphagia, and loss of postural reflexes; (ii) progressive cognitive disturbance culminating in dementia; and (iii) various behavioral disturbances including depression, anxiety, apathy, obsessive–compulsive behaviors, outbursts, and occasionally delusions or psychosis. A cognitive or behavioral syndrome is often the first manifestation, a fact that is often clearer in retrospect. Weight loss is another common symptom [18]. Though chorea is only a small part of motor dysfunction in HD, it remains its most recognizable feature. Chorea often begins as fleeting, suppressible random “fidgety” movements, seen best in the distal extremities. With time, chorea becomes insuppressible and more overt, involving larger and more proximal muscles. Chorea might incorporate fragments of complex purposeful movements such as straightening one’s hair. Motor impersistence is often seen in chorea. The “flycatcher’s tongue,” for example, describes a difficulty in keeping the tongue protruded beyond the lips. Most patients with chorea are not aware of the extent of their involuntary movements; some deny it altogether. Others report that the movements are embarrassing, exhausting, or injurious. In many patients, worsening chorea develops the sinuous, writhing quality known as athetosis. The combination of athetosis and the more rapid movements of chorea is termed choreoathetosis. Severe chorea–choreoathetosis exhibits violent flinging of the limbs or trunk and can be accompanied by traumatic injuries. Saccadic eye movement abnormalities occur early and persist throughout the disease. Saccades are slow to initiate, often requiring a head movement or a blink to break fixation. Saccadic velocities often become slow as the disease progresses [19]. Patients usually do not complain of associated visual problems. Ataxia, manifested by dysmetria and dysrhythmia, is common in HD, especially as disease advances. Ataxia might be apparent in speech, finger–nose, and heel–knee–shin testing and a broader-based gait with impaired tandem walking. Dystonia, a more sustained posturing or twisting, is common in HD. Bradykinesia refers to slower and reduced amplitude of movement: for example, diminished facial expression, reduced spontaneous gesturing, reduced finger tap speed and size, reduced arm swing, and small steps. Well before HD manifests, gene-positive individuals show increased variability of voluntary motor measures such as finger tap and tongue protrusion force [20]. There is considerable motor heterogeneity from patient to patient. In general, younger-onset patients are more likely to present with bradykinesia, rigidity, and dystonia; juvenile HD patients may lack chorea altogether and look more like they have Parkinson’s disease (PD) (known as the “Westphal variant” of HD). These individuals have a relatively high prevalence of epilepsy [21]. Even within a given HD patient’s disease course, the motor abnormalities evolve across a continuum: chorea early in the disease often progresses to superimposed dystonia as the disease advances [22], culminating often in striking bradykinesia, rigidity, and poor postural reflexes in late stages. Progressive motor failure is a major cause of life-ending complications; falls and serious injuries become increasingly common. Weight loss in HD occurs even early in the disease, independent of chorea, and might represent a metabolic or homeostatic alteration due to the disease [23]; dysphagia in late HD only exacerbates this problem. Dementia is sometimes an underappreciated facet of HD and especially serious because it strikes in the prime of life. Unlike elderly demented adults, HD patients often lack adult children who can assist with planning and care. Marriages and families are disrupted, and children lose an effective parent. With increasing difficulty at work, years of income are lost. A recent large-scale prospective observational analysis of premanifest persons [24] showed declines in several measures, including working memory, attention, and verbal fluency, consistent with prior studies. These deficits were worse for subjects approaching their expected motor onset. Prediagnosis cognitive impairment, as measured via neuropsychological testing, may serve as an important outcome measure in future clinical trials of potential preventive agents. By the time of diagnosis, most subjects with HD have cognitive impairment clearly evident on neuropsychological testing. Patients typically have difficulty with multitasking, focus, short-term memory, and learning new skills, although these problems are often first noted by family members. Early in the disease, these difficulties are usually not sufficient to impede most activities of daily living, but those with cognitively demanding jobs often find work increasingly difficult. Over many years, cognitive impairment eventually progresses to frank dementia in most patients with HD. Unlike Alzheimer dementia, HD dementia is largely “subcortical,” marked by slow thought processes, executive dysfunction, and problems with attention and sequencing [25, 26]. Episodic memory, though impaired, is relatively well preserved when compared to Alzheimer’s disease (AD), as is language function. The MMSE is ineffective as a screening instrument in HD; the MoCA is considerably more sensitive in this population [27] as it contains more items evaluating executive function and attention. The Luria test (fist–edge–palm) requires good motor sequencing and executive function, so it is impaired particularly early in HD. Individuals with HD often show striking lack of insight into their own cognitive and motor symptoms, even when these are obvious to others [28]. This might reflect dysfunction of striatal neurons receiving prominent frontal lobe inputs. For many HD patients and their families, behavioral problems are the most vexing. These range from affective illness to anxiety disorders to delusional behavior and rarely hallucinations [29, 30]. Psychiatric features and their severity vary tremendously and do not correlate with chorea or dementia [29]. Psychiatric problems are especially problematic in juvenile-onset cases [31]. Most patients experience some behavioral symptoms prior to their diagnosis [32–34]; most common are depression, obsessive–compulsive behaviors, irritability, and outbursts [32, 33]. Personality changes may occur for years prior to the diagnosis, though this may be apparent to families only in retrospect. Up to 50% of patients are depressed at some point in the disease [35]. Depression often responds very well to treatment, typically SSRI antidepressants. Apathy is also fairly common, though more difficult to treat. Compulsive behaviors in HD may resemble the cognitive rigidity and perseveration typical of frontal lobe disorders and probably reflect striatal dysfunction. There is a high rate of suicide in gene-positive individuals, both after diagnosis and prediagnosis [36, 37]. Factors increasing suicide risk include being single, lacking children, living alone, being depressed, and showing manifest HD signs [36]. There is a remarkable range of ages of onset and symptom features in HD. CAG repeat length is a major factor, correlating inversely with age of onset [38–41]. For the more common smaller repeat lengths (<45), however, there is a much larger variance in age of onset. Other genetic and environmental factors, yet unknown, must also contribute to age of onset. Repeat length also influences motor phenotype, as early-onset disease is more likely to present with prominent dystonia and bradykinesia, though this may reflect the impact of the mutant allele on developing brains. It is unclear whether repeat length influences the rate of disease progression; whereas some studies have not [39], other studies have [42] found correlations. Age may have been a confounder; a more recent analysis showed a stronger correlation between longer repeats and faster decline after adjusting for age [43]. Familial aggregation of certain symptoms (e.g., psychosis) occurs in HD, and this likely reflects genetic modifiers. Mimics of HD are seen occasionally in clinical practice [44, 45]. The differential diagnosis for chorea alone is extensive and includes acquired entities such as hyperthyroidism, polycythemia vera, lupus, antiphospholipid antibody syndrome, HIV/AIDS, poststreptococcal chorea, anticholinergic and stimulant drug effects, and levodopa-induced dyskinesias in PD. Most such entities can be excluded by history, physical examination, or simple blood tests. More difficult to distinguish are a few inherited neurodegenerative diseases that might resemble HD. These include dominant disorders such as the polyglutamine diseases, SCA17 and DRPLA; Huntington’s disease-like 1 (HDL1), due to a mutation in the gene for the prion protein; HDL2, which is another expanded repeat disorder that closely resembles HD and is quite rare; and the neurodegeneration with brain iron accumulation (NBIA) disease spectrum. Recessive and X-linked disorders in the neuroacanthocytosis family might mimic HD. Molecular diagnoses are available for all these diseases. Because it is treatable, Wilson’s disease must be considered in any nonelderly person (<50 years of age) presenting with a movement and cognitive disorder, though a chorea-dominant phenotype is uncommon. Because HD is the most common progressive choreiform disorder, the simplest strategy is to first test for the HD mutation. This should be performed even if there is no apparent family history; there may be a new mutation, early death, or missing information on a parent, nonpaternity, or undisclosed adoption. If HD is excluded, more extensive testing would then be needed. Establishing an accurate diagnosis is crucial for both patient and family; the implications are vastly different for a dominant versus a recessive disorder, and an accurate diagnosis is required for presymptomatic testing of unaffected adult family members over the age of 18. If there is a confirmed family history of HD (i.e., an affected parent or sibling with an established genetic diagnosis), mutation testing is generally not required in a patient presenting with motor abnormalities that are unequivocally characteristic of HD. A note of caution, however, is that mild motor anomalies, including subtle chorea, cannot be assumed to be diagnostic of HD simply on the basis of a known family history. Even mutation-negative relatives sometimes appear hyperkinetic, perhaps from behavioral imprinting. Currently, neuroimaging is of limited value in the clinical diagnosis of HD. Individuals with many years of manifest HD typically demonstrate obvious caudate atrophy on CT or MRI, altering the contour of and enlarging the lateral ventricles. Volumetric MRI analyses, performed in ongoing large prospective studies of at-risk subjects, reveal that the most clearly measurable and progressive atrophy affects the striatum and global cerebral white matter [46, 47] (see Figure 8.1). These changes occur well before the earliest typical motor features. Cortical atrophy also occurs in asymptomatic subjects [20, 49, 50], changing quantitatively over relatively short intervals (2–3 years), consistent with histopathologic findings of early neocortical degeneration [51]. There is tremendous interest in developing such MRI analyses as biomarkers for use in future clinical trials with premanifest subjects. Figure 8.1 Coronal T1-weighted brain MRI in Huntington’s disease. (a) Healthy control. (b) HD carrier with early-stage (stage 2) disease. MRI shows caudate (white arrow) and putaminal (black arrow) atrophy showing that there is enlargement of the lateral ventricles due to partial degeneration of the caudate nucleus. Source: Mascalchi [48] (TBC). Reproduced with permission of Radiological Society of North America. PET studies have demonstrated early declines in striatal neurotransmitter markers, particularly dopamine receptors, that occur even in preclinical HD [52–54]. The HTT gene on chromosome 4 encodes the protein huntingtin [55], a very large protein that is expressed widely in the CNS and in other tissues. In neurons, it is found largely in somatodendritic and axonal cytoplasm and interacts with many other proteins. Huntingtin is essential for early neuronal development, but its precise functions in adults are unclear. It appears to be important in processes as diverse as protein and vesicular transport, signaling, transcriptional regulation, and apoptosis [56]. How mutant huntingtin initiates neurodegeneration is still unknown, though a toxic gain-of-function mechanism occurring primarily at the protein level is very likely. There is tremendous interest in identifying reliable biomarkers to pinpoint HD onset, track its progression, and determine response to treatment. Existing markers are useful but clearly not sufficient; the best known is the Unified Huntington’s Disease Rating Scale (UHDRS) [57]. This standardized clinical instrument has four major components: motor, cognition, behavior, and functional ability; it has been widely used in HD clinical trials and observational studies. A subset of the full UHDRS, perhaps supplemented with specific cognitive assessments, might someday constitute a straightforward battery of tests that reliably measures disease onset and progression. Other candidate biomarkers might directly measure an aspect of HD pathophysiology; examples include changes in brain imaging (reviewed earlier), metabolic/proteomic profiles [58, 59], or gene expression changes [60].
Repeat expansion diseases and dementia
Introduction
HD
Nosology
History/nomenclature
Epidemiology
Genetic epidemiology
Case presentation
Clinical features
Overview and natural history
Motor disorder
Cognitive disorder
Psychiatric disorder
Clinical heterogeneity
Differential diagnosis
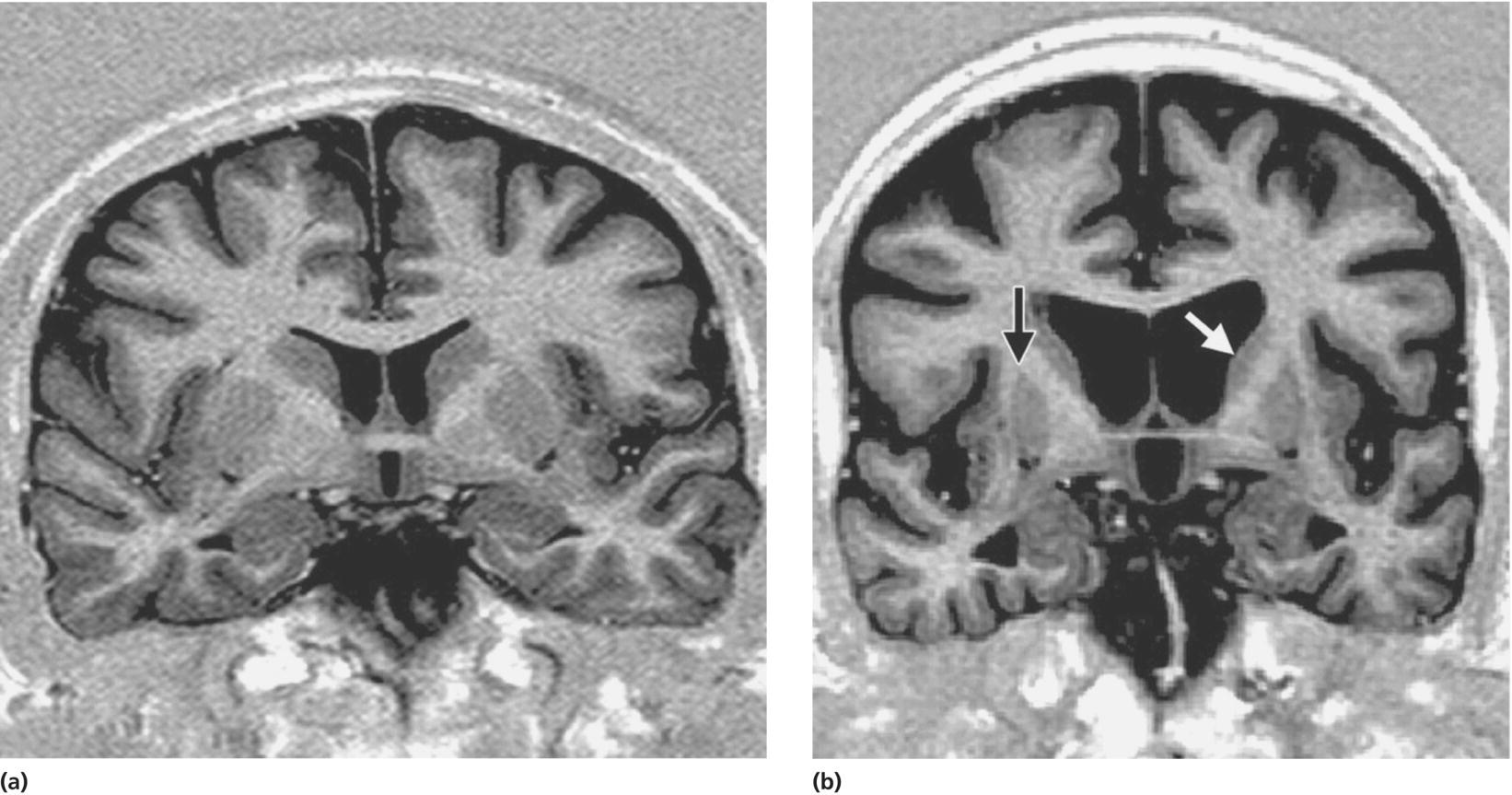
Neuroimaging findings
Pathophysiology and pathology
The HTT gene
Biomarkers
Neuropathology
Gross CNS pathology
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


