CHAPTER 9 Leonel T. Takada1 and Michael D. Geschwind2 1 University of Sao Paulo Medical School, Sao Paulo, Brazil 2 University of California, San Francisco, San Francisco, CA, USA Rapidly progressive dementias (RPDs) often are defined as disorders that cause progressive cognitive decline (usually with other signs and/or symptoms), in which the interval between the first symptom to the onset of dementia is typically over weeks to months, but almost always less than 2 years [1–6]. Some have defined rapidly progressive neurodegenerative diseases as those in which survival is less than 3 years [3, 4] as opposed to the 9–12 years typical for most neurodegenerative diseases, such as Alzheimer’s disease (AD) and frontotemporal dementia (FTD) [7]. RPDs might also be thought of as conditions in which Jakob–Creutzfeldt disease (CJD), the prototypical RPD, is considered in the differential diagnosis. CJD is the most frequent form of prion disease (PrD) and one of the most common etiologies of RPD [1, 3, 8, 9]. CJD and other PrDs therefore will be the primary focus of this chapter. RPDs can also be caused by a myriad of conditions, many of which are treatable, and thus the evaluation of RPDs is challenging not only for the variety of differential diagnoses but also for the sense of urgency to find a reversible or treatable cause. Potentially reversible conditions that cause RPDs are not infrequent, even at PrD centers [1, 8]. In our UCSF RPD program, whereas PrDs were diagnosed in 62% of the suspected CJD referrals, about 16% had potentially treatable causes (autoimmune, infectious, psychiatric, neoplastic/paraneoplastic, or toxic-metabolic) [1]. At the US National Prion Disease Pathology Surveillance Center (NPDPSC), although 68% of the 1106 autopsied cases were positive for PrDs, 6.1% had potentially treatable conditions (immune mediated, infectious, toxic-metabolic, or neoplastic) [8]. Many of the possible differential diagnoses for RPDs have been discussed in other chapters within this volume; thus, in this chapter, we will briefly discuss the diagnoses not previously covered and provide an algorithm for the clinical evaluation and diagnosis of RPDs. PrDs are a group of neurodegenerative disorders caused by infectious proteins called prions. The term prion is derived from “proteinaceous infectious particle” and was coined by Stanley Prusiner [10], the 1997 Nobel Prize in Physiology or Medicine laureate for his work on identifying it as the causative agent of transmissible spongiform encephalopathies (TSE). PrD may occur in many species (viz., scrapie in sheep and goat, bovine spongiform encephalopathy [BSE] in cattle, chronic wasting disease in cervids, transmissible mink encephalopathy in minks, feline spongiform encephalopathy in cats, and exotic ungulate encephalopathy in greater kudu, nyala, and oryx) [11], but this chapter will focus on the human forms—for which we will use the single eponym, CJD, when referring to human prion diseases (hPrDs) in general. CJD was first described in 1921 by the German neurologist and neuropathologist Alfons Jakob [12, 13], who felt his five cases resembled the one published by Hans Creutzfeldt 1 year prior. It is now known that Creutzfeldt’s case was not PrD (only two of Jakob’s cases are what we now consider CJD) [14]. For years, the disease was referred to as Jakob–Creutzfeldt or Jakob’s disease. A prominent PrD researcher, C.J. Gibbs preferred to have the eponym match his own initials, so he began to call the disease Creutzfeldt–Jakob disease (CJD) [15], and the name has largely stuck, although it is historically inaccurate. In this chapter, we will use the eponym Jakob–Creutzfeldt disease but still keep the acronym CJD to avoid often and erroneous associations with the JC virus, causative agent of progressive multifocal leukoencephalopathy. Currently, hPrDs are classified in three groups: sporadic (85–90% of cases), genetic (10–15%), and acquired (1–3%) [16, 17]. The sporadic forms are called sporadic CJD (sCJD). The genetic forms historically are subdivided into three categories—familial CJD (fCJD), Gerstmann–Sträussler–Scheinker (GSS) disease, and fatal familial insomnia (FFI)—and the acquired forms include kuru, iatrogenic CJD (iCJD), and variant CJD (vCJD). The incidence (which in this case is comparable to mortality rate, because of the short disease duration in CJD) of sCJD is about 1–1.5 per million per year in most developed countries, with some variability from year to year and between countries [17, 18]. The mortality of genetic prion diseases (gPrDs) was calculated in a pooled data from the European countries, Canada, and Australia to be 0.17 per million per year, but with a wide range (0.01–1.07) [17]. The peak age of onset of sCJD occurs around a unimodal relatively narrow peak of about 68 years [19], with an age-of-onset range of 12–98 years [20, 21] (Geschwind, personal communication). CJD is rare in individuals younger than 30 years, and most of those cases are either acquired or genetic [18, 22]. There is no gender preference in sCJD, although there might be a female preponderance, possibly due to females living longer than men, creating a survival bias [18]. In the United States, an incidence 2.5 times higher in Caucasians compared to African Americans was found [18], but other interethnic comparison data is still lacking. Gajdusek and colleagues were the first to demonstrate that human spongiform encephalopathies were transmissible [23] and, for a long time, slow viruses were thought to be the infective agents. Alper, Pattison, Griffith, and others postulated that the agent causing scrapie did not contain nucleic acid and actually might be a protein [24–27]. Prusiner and colleagues’ groundbreaking work helped prove the concept that proteins could act as infections agents [10]. It is now generally accepted that PrDs are caused by the propagation of abnormally conformed infectious proteins called prions (also named PrPSc, for prion-related protein in which “Sc” comes from scrapie). The normal cellular prion-related protein (PrPC, in which “C” stands for the normal cellular form) is a membrane-bound protein that is predominantly expressed in nervous tissue, but its physiologic function is not entirely known (although it probably plays a role in neuronal development and function) [28, 29]. Interestingly, recently, PrPC has also been implicated in the metabolism of the amyloid-β peptide, one of the main proteins involved in AD pathogenesis [30]. Prion infectivity occurs through a mechanism in which the pathogenic PrPSc act as a template to convert PrPC into PrPSc [11, 31], such that when PrPSc, which has mostly beta-pleated sheet structure, comes in contact with PrPC, which has mostly alpha-helical structure, PrPC is misfolded into the proteinase K-resistant PrPSc. This new PrPSc then becomes a template for the conversion of existing PrPC, initiating an exponential, cascade reaction, which leads to neuronal injury and death. PrDs are unique, as they can occur as sporadic, genetic, and infectious diseases [11]. Though the initial pathogenic step is not clear, sporadic PrD is thought to occur by spontaneous folding of PrPC into PrPSc (or maybe through a somatic mutation in the PRNP gene) [10, 32]. In gPrD, it is believed that mutations in PRNP gene make PrPC more susceptible to changing conformation into PrPSc [33]. In the orally acquired forms of PrD (such as kuru and vCJD), a currently accepted mechanism of neuroinvasion starts with the uptake of prions through the intestinal epithelium. Prions then accumulate in the lymphoid tissue before being transported via sympathetic and parasympathetic nerves to the central nervous system (CNS) [34]. Human PrPSc often is subclassified based on the fragment properties resulting from digestion by proteinase K and running on a Western blot into type 1 and type 2 [35]. Type 1 PrPSc fragments are approximately 21 kDa and type 2 are approximately 19 kDa in unglycosylated forms. Brains of patients with sCJD can have either or both types [36]. This has implications on clinical and pathological features, as will be briefly discussed below. In humans, the prion-related protein gene (PRNP) is located in chromosome 20p13 and is considerably conserved among mammals. Some interspecies sequence variation occurs, however, and might be responsible for the species barrier seen in some experiments on PrD transmissibility, as the differences between the infectious PrPSc and the host’s PrPC appear to influence infectivity and incubation period [32]. For example, the bank vole is very susceptible to prions from many species. Transgenic mice containing bank vole Prnp are much more susceptible than wild-type mice to prions from other species [37]. Only mutations in PRNP are known to cause gPrD [38, 39], and there are currently more than 40 known mutations in this gene [32, 40]. The majority of mutations are missense mutations, with the most common mutations being E200K (the most common worldwide), D178N, P102L, and V210I [41–43]. Stop codons, insertions (usually octapeptide repeat insertion (OPRI) mutations), and deletions of PRNP are less common causes of gPrD. The pattern of inheritance of gPrD is autosomal dominant and penetrance is usually 100%. Some mutations causing gPrD [44–46] do not have 100% penetrance, however [42]. Among E200K mutation carriers, for example, penetrance appears to be age dependent (i.e., 1% at age 40, close to 100% after the ninth decade of life); as such, it is not uncommon to find older asymptomatic mutation carriers [47, 48]. It is also known that variations (polymorphisms) in PRNP influence an individual’s susceptibility to develop disease. The most acknowledged and important polymorphism is located in codon 129, which can have either valine (V) or methionine (M) as alleles (and so the three possible combinations are MM, MV, and VV). There is a clear overrepresentation of homozygotes (MM or VV) among PrD patients. Whereas in a normal Caucasian population about 50% are heterozygous (MV), 40% are MM, and <10% VV [49]; in every form of PrD, more than 65% of the patients are either MM or VV [17]. The particularities of codon 129 polymorphisms in each form of PrD will be discussed in the appropriate sections. The clinical presentation of sCJD is highly variable. In most cases, onset is subacute, although in a few cases onset may be acute or stroke-like [50]. In Mr. F’s case, the first symptom was unspecific sensory changes, followed by cerebellar symptomatology. Cerebellar symptoms are the initial manifestation in about 20% of cases, as are constitutional symptoms (such as dizziness, headaches, sleep or eating changes, or fatigue) and behavioral symptoms (e.g., depression, irritability) [51]. In about 40% of patients, the initial symptom is cognitive (most commonly presenting as memory problems, executive dysfunction, and/or language impairment) [19, 51]. Visual changes (blurred vision, diplopia, oculomotor changes, visual hallucinations) occur as a first symptom in 10–15% of cases [19, 51, 52]. Extrapyramidal (i.e., parkinsonism, dystonia, myoclonus, chorea) and pyramidal motor symptoms are less frequently seen as the early or presenting manifestation but are more likely to be seen as the disease progresses. Parkinsonism in sCJD might manifest with supranuclear gaze palsy, early gait problems, and/or alien limb, sometimes resembling atypical parkinsonism [53]. Dystonia is rare in AD and dementia with Lewy bodies (DLB) but has been reported to be seen in as many as 20% of sCJD cases [54]. Myoclonus is rarely a presenting symptom but is seen during the clinical course in almost 90% of cases per one large retrospective study [19]. Myoclonus usually starts in a limb and eventually becomes generalized; sometimes, it is associated with periodic sharp wave complexes (PSWCs) on EEG [53]. Seizures (or even status epilepticus) are rarely described as an initial manifestation of sCJD but can occur, usually later, in the disease in a minority (~8–9%) of cases [19, 52, 55]. Clinically evident peripheral neuropathy (including cranial neuropathy) is uncommon in sCJD [56], even though PrP deposits may occasionally be found in the peripheral nerves [56, 57]; certain gPrDs, such as E200K among Libyan Jews, more commonly have neuropathy [58–60]. Symptoms usually follow a rapidly progressive course, and different manifestations are added to the symptomatology throughout the course of the disease. In Mr. F’s case, cerebellar ataxia was soon followed by cognitive and behavioral changes, and the neurological examination revealed subtle extrapyramidal signs. The common final pathway of sCJD in most cases is the development of akinetic mutism, the terminal extrapyramidal manifestation [53]. Patients with sCJD and other PrDs usually die from aspiration pneumonia. Median duration of disease is reported to be around 4–6 months (mean 7 months), and death occurs within 1 year in 90% of cases, with another 5% of patients dying in the second year of disease [19, 61]. Younger age of onset, female gender, and heterozygosity at PRNP codon 129 have been associated with longer survival in sCJD [61]. sCJD has been divided into six molecular subtypes based on the polymorphisms at codon 129 in the prion gene (MM, MV, or VV) and the type of protease-resistant prion (type 1 or 2) [35]. This classification, to some extent, separates sCJD cases based on their clinicopathological features. The MM1 and MV1 variants are the most frequent type (60–70% of cases) and are characterized by sCJD with a faster course, with a reported median survival of about 4 months. The VV2 type is characterized by ataxia with a later age of onset and short disease duration (about 6.5 months); the MV2 type is similar to the VV2 but has longer disease duration (17.1 vs. 6.5 months). The VV1 type is the less frequent variant and associated with the earliest age of onset. The MM2 type is further divided into cortical and thalamic type; the MM2 thalamic type is considered by some as the sporadic form of fatal insomnia. Since this classification scheme was established, it later was reported that type 1 and 2 prions often coexist in the same patient, so this classification scheme will need some revision. Preliminary analysis suggested that MM1/2 type phenotypically falls in between MM1 and MM2 subtypes [35, 36, 62, 63]. Amidst the great variability in clinical presentation, a few sCJD variants are recognized: the Heidenhain variant (less than 10% of sCJD cases; it is characterized by visual symptoms at presentation, such as visual hallucinations or distortions, cortical visual deficits, and/or oculomotor impairment; mostly MM1 [48, 64]), the Brownell–Oppenheimer variant (with ataxia as the presenting and dominant symptom, and lack of EEG PSWCs and deep nuclei hyperintensities [64]), a thalamic variant (sometimes referred to as sporadic fatal insomnia [sFI]; linked to MM2 [65, 66]), the panencephalopathic form (with significant or primary involvement of white matter; described primarily in Japan and only rarely seen in Caucasians [see following text, in neuroimaging] [67, 68]), and an amyotrophic form (with motor neuron disease findings) [69]. More recently, a new form of PrD was described, based on the finding that in a group of cases with similar clinical and neuropathological features, the PrPSc was more sensitive to proteinase K digestion. Those cases were termed as having variable proteinase-sensitive proteinopathy (VPSP) and are clinically characterized by having aphasia, ataxia, and parkinsonism as prominent manifestations and a longer disease course than sCJD [63, 70, 71]. The most commonly used diagnostic criteria are the ones proposed by the World Health Organization (WHO) in 1998 [72]. Those criteria do not take into consideration MRI findings (which are the most sensitive diagnostic test for PrD), and so, more recently, two other criteria have been published (see Table 9.1 for a review of all three). Table 9.1 Diagnostic criteria for sporadic Jakob–Creutzfeldt disease. * Clinical criteria as originally printed in [188] were erroneous, leaving out myoclonus and putting dementia as one of the four possible features. † Higher focal cortical signs include such findings or symptoms as apraxia, neglect, acalculia, aphasia, etc. CSF, cerebrospinal fluid; DWI, diffusion-weighted imaging; EEG, electroencephalogram; FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging; PSWCs, periodic sharp wave complexes. Diagnostic criteria for sCJD are developed primarily for research purposes and aim to obtain the highest accuracy in predicting neuropathologically confirmed CJD. Because of that, the criteria are not particularly sensitive early in the disease course, and most cases will only fulfill diagnostic criteria later in the illness (e.g., akinetic mutism is one of the clinical criteria, despite being a late occurrence). The UCSF criteria for sCJD were first proposed in 2007, and they modified the WHO symptom criteria by substituting MRI for the CSF 14-3-3 protein [2, 73], separating visual and cerebellar symptoms from one another and adding the symptom of higher focal cortical signs. The UCSF CJD MRI criteria were updated in 2011 and are detailed in Table 9.2 [74]. Table 9.2 UCSF 2011 MRI criteria for sCJD. Source: Vitali et al. [74]. Reproduced with permission of Wolters Kluwer Health, Inc. Supportive for cortical involvement: ADC, apparent diffusion coefficient; CJD, Creutzfeldt–Jakob disease; DWI, diffusion-weighted imaging; FLAIR, fluid-attenuated inversion recovery; sCJD, sporadic Creutzfeldt–Jakob disease Brain MRI is currently the most accurate method for the diagnosis of sCJD, with approximate sensitivity of 92–96% and specificity of 92–94% using diffusion-weighted imaging (DWI) [74, 75]. MRI changes can also be seen very early in the disease course (even though in some cases MRI abnormalities will only appear with repeated MRIs) [75], further enhancing its value for the diagnosis. MRI used for the diagnosis of CJD should always include DWI and apparent diffusion coefficient (ADC) sequences, as DWI findings are far more sensitive than FLAIR and/or T2 sequence abnormalities [74, 75]. There are three major patterns of DWI MRI hyperintensities in sCJD (Figure 9.2): cortical and deep nuclei (68% of cases), predominantly neocortical (24%), and predominantly deep nuclei (primarily the striatum; with or without thalamic changes—the pattern seen in Mr. F’s case, Case 1) (5%) [74, 76]. The molecular classification of PrD seems to affect the pattern of MRI involvement [77]. Cortical hyperintensities (or cortical ribboning) can be seen in almost any neocortical region but with relative sparing of the precentral cortex [74]. Figure 9.2b shows an example of a predominantly cortical DWI in sCJD. Deep nuclei hyperintensities usually involve the striatum, often with an anterior-posterior gradient (i.e., the anterior caudate being more hyperintense than the posterior putamen) [74]. Involvement on MRI may be bilateral and symmetric, bilateral but asymmetric, and even completely unilateral. Limbic (i.e., insula, anterior cingulate, hippocampus) hyperintensities may be seen as an additional finding in up to 90% of sCJD cases (with the caveat that these regions are more frequently associated with artifacts and/or false-positive abnormalities), but predominant or isolated limbic abnormalities generally are not characteristic of sCJD and should make one consider a nonprion diagnosis (particularly infectious or autoimmune encephalitis and seizures) [74]. Whenever an area of hyperintensity is questioned to be false positive, adding coronal and sagittal acquisitioned images to the evaluation, as well as searching for correspondent ADC map hypointensities, may be helpful [74]. Figure 9.2 Axial brain MRI in sporadic CJD and variant CJD. (a-c) Each show a FLAIR, DWI and ADC sequences, whereas d shows only a FLAIR and DWI sequence. (a) Neocortical (solid arrow), limbic (dashed arrow) and subcortical (dotted arrow) DWI and FLAIR hyperintensities with corresponding ADC hypointensities in sporadic CJD. (b) Neocortical (solid arrow) and limbic (dashed arrow) DWI hyperintensities with corresponding ADC hypointensities in sporadic CJD. (c) Subcortical (dotted arrows) DWI and FLAIR hyperintensities, with corresponding ADC hypointensities in sporadic CJD. (d) Pulvinar sign (arrow) in variant CJD. ADC, apparent diffusion coefficient; CJD, Creutzfeldt–Jakob disease; DWI, diffusion-weighted imaging; FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging. Source: Adapted from Vitali et al. [74, 189]. Reproduced with permission of Wolters Kluwer Health, Inc. The DWI hyperintensities have a pattern of water diffusion restriction (probably caused by vacuolation [78]), with corresponding hypointensities in the ADC maps. The ADC hypointensities are more easily identified in the deep nuclei but may also be found in cortical regions [74]. Cortical ribboning on DWI can also be seen in viral encephalitis, seizures, status epilepticus, hypo- and hyperglycemia (often with seizures), Wernicke’s encephalopathy due to thiamine deficiency, and acute stroke (though usually associated with concomitant white matter abnormalities). Striatal or thalamic DWI hyperintensities with ADC hypointensities have also been described in extrapontine myelinolysis, Wilson’s disease, Wernicke’s encephalopathy, Bartonella infection, and hyperglycemia with seizures (as reviewed by Vitali et al. [74]) [79–81]. Mitochondrial disease, vasculitis, acute phase of hypoxic ischemic encephalopathy, posterior reversible leukoencephalopathy, and lymphoma may also be considered in the differential diagnosis of DWI hyperintensities [75, 82]. White matter abnormalities are typically absent in sCJD, except in the rare panencephalopathic form of CJD, which is associated with major white matter hyperintensities in T2-weighted MRI [83]. Many feel that the panencephalopathic form of sCJD merely occurs in patients with prolonged courses, often due to life-extending measures such as feeding tubes or intubation, and is due to Wallerian degeneration [67]. With the progression of disease, particularly in patients with disease duration of over 1 year, as atrophy progresses, DWI hyperintensities might fade away and be absent on later MRI scans [74]. There are no currently available clinical blood tests for the diagnosis of CJD. Blood tests, however, are always necessary to exclude other causes of RPD, as will be discussed in the following. General cerebrospinal fluid (CSF) analysis is typically normal in sCJD, except mildly elevated protein (typically less than 75 mg/dl) is not uncommon. Although elevated protein (>75, <100 mg/dl), pleocytosis (>5 WBC cells), elevated IgG index, or the presence of oligoclonal bands rarely do occur in sCJD [84], their presence should lead to considering other conditions, particularly infectious or autoimmune disorders. The most common CSF markers used in clinical practice are 14-3-3 protein, total tau (t-tau), and neuron-specific enolase (NSE) (S100β is used primarily in the United Kingdom [85]), but due to great variability in accuracy across studies, the clinical value of each marker is still not entirely clear. A large European study has found the sensitivity and specificity of the 14-3-3 to be 85 and 84%, t-tau (cutoff >1300 pg/ml) 86 and 88%, NSE 73 and 95%, and S100β 82 and 76%, respectively [86], and CSF markers collected later in the disease course seemed to be more sensitive than those collected earlier [87]. Among these three more common biomarkers, t-tau might be the best CSF diagnostic marker for sCJD, although there is still no complete agreement over its cutoff value, which tends to vary between 1150 and 1300 pg/ml. Combining markers also seem to increase their diagnostic value [85, 86]. The 14-3-3 protein is a nonspecific marker for neuronal injury and can be increased in non-PrD, such as cerebrovascular disease, metabolic and hypoxic encephalopathies, brain metastases and CNS infections, or even other neurodegenerative dementias [85, 88]. Overall, the 14-3-3 is not a very specific test; one large study found it to have about 40% specificity for CJD [63]. Among PrDs, 14-3-3 protein is not consistently elevated in genetic and iatrogenic forms [89]. A new assay for detecting prions, called real-time quaking-induced conversion (RT-QuIC), can detect very minute amounts of PrPSc by using in vitro conversion of PrPC as a substrate [90]. The test is based on a combination of two methods for prion detection—a PCR-like method of amplifying prions called protein misfolding cyclic amplification (PMCA) [91] and an amyloid seeding assay [92]. The sensitivity and specificity of this assay in CSF for sCJD appear to be around 87 and 98%, respectively, based on preliminary studies [93, 94]. A typical EEG in sCJD has sharp, or triphasic, waves (PSWCs) occurring about once every second. This EEG finding, however, is found in only about two-thirds of sCJD patients and usually only after serial EEGs and/or not until later stages of the illness [95]. Often, the only finding is focal or generalized slowing. PSWCs are relatively specific, but they can also be seen in other conditions, including toxic-metabolic and anoxic encephalopathies, progressive multifocal leukoencephalopathy, AD, Lewy body dementia, and Hashimoto’s encephalopathy [96, 97]. Mild atrophy usually is the only gross neuropathological finding in sCJD brains, and it is not always present. The typical neuropathological findings are neuronal loss, gliosis, and vacuolation (or spongiform changes), without inflammatory signs (Figure 9.3). Current diagnostic criteria for definite sCJD also require positive PrPSc tissue immunoreactivity [98, 99]. PrP amyloid plaques (or kuru plaques) are found in 5–10% of sCJD cases, particularly MV2 [65, 99]. These plaques are more characteristic of GSS, as will be discussed in the following section. Figure 9.3 Neuropathological findings in prion diseases. (a) In sporadic CJD, some brain areas may have no (hippocampal end plate, left), mild (subiculum, middle), or severe (temporal cortex, right) spongiform change. Hematoxylin and eosin (H&E) stain. (b) Cortical sections immunostained for PrPSc in sporadic CJD: synaptic (left), patchy/perivacuolar (middle), or plaque type (right) patterns of PrPSc deposition. (c) Large kuru-type plaque, H&E stain. (d) Typical “florid” plaques in vCJD, H&E stain. Source: Adapted from Budka [98]. Reproduced with permission of Oxford University Press. gPrDs historically have been divided according to clinical and pathological characteristics into three forms: fCJD, GSS, and FFI. Identifying the prion gene, PRNP, has helped classify gPrDs more precisely by specific mutations, which include mostly point mutations, but also insertions (octapeptide repeats) and deletions [101]. A caveat to classifying patients by their PRNP mutation is that a single PRNP mutation can be associated with different phenotypes (and great variability even within a single family) [102]. Some of this variability depends on the codon 129 polymorphism but other unidentified factors as well. Another fundamental issue is that gPrDs are sometimes referred to as familial, but considering that up to 60% of the gPrD cases do not have a positive family history [42], the term “familial” can be misleading. We prefer not to use the term fCJD, as many patients with gCJD or gPrD, a family history of PrD is not known; this can be due to several reasons, including reduced penetrance or misdiagnosis [42, 46]. In those cases of negative family history, often there is a history of family members being (mis) diagnosed with more common neurodegenerative diseases such as AD or Parkinson’s disease [42]. Other possibilities are incomplete or age-dependent penetrance [46, 103] (such as in case 1’s, Mr. F’s, brother and possibly his father, whose death from stroke may have occurred before he would develop CJD symptoms) or incomplete family history (the disease may even be kept secret from younger members of a family; M. Geschwind, personal experience). The clinical features of gCJD are highly variable, and inter- and intrafamiliar variations may be seen (not only the mutation but also codon 129 polymorphism may affect presentation) [65]. As a group and in comparison to sCJD, gCJD is associated with a younger age of onset (typically <55 years; but onset may occur as late as the ninth decade) and longer clinical course [41, 48, 61]. Also, ancillary testing including CSF, EEG, and MRI may not be as sensitive or specific as in sCJD [41, 65, 104]. The E200K mutation is the most frequent PRNP mutation worldwide, and higher frequency of this mutation has been found among Libyan Jews and in Slovakia [105]. The clinical features (and neuropathological features) are highly variable, but in general comparable to sCJD, including age of onset and duration of disease [41]. Dementia and ataxia are the most frequently described symptoms, but vertical gaze palsy, polyneuropathy, and sleep changes have also been reported [60]. Mr. D (Case 2) had the prototypical GSS, which typically presents as a subacute progressive ataxic and/or parkinsonian disorder with later onset of cognitive impairment, and onset most commonly occurs in the fourth to sixth decades (but can occur as early as the twenties [41]). Because it is usually slower than sCJD or many other gPrDs, often with duration of about 5 years (range 3–8 or more years), the differential diagnoses of GSS are ataxic and/or parkinsonian conditions, such as multiple system atrophy, other atypical parkinsonian disorders, idiopathic Parkinson’s disease, as well as ataxic disorders such as spinocerebellar ataxias or Huntington’s disease. Pyramidal signs may also be found, and lower limb dysesthesia and areflexia may be other associated clinical features, especially in the P102L mutation [106, 107]. There is considerable phenotypic variability within and between mutations and families, and some cases may not even have ataxia as a main characteristic and rather present with early dementia and/or behavioral abnormalities [41, 48]. At least 15 PRNP mutations have been shown to cause GSS [102]. EEG in most cases does not show typical CJD findings, and CSF protein 14-3-3 is increased in about 50% of cases [42]. MRI scans are usually normal, and some degree of brain and cerebellar atrophy may be seen with the progression of disease [106]. Cortical ribboning or deep nuclei (striatal or thalamic) hyperintensities on T2/FLAIR or DWI are uncommon findings in GSS but have been reported [106, 108, 109]. Our own study found that limbic DWI or FLAIR hyperintensities can be found in some cases, but it was not clear that these were true diffusion restricted [74]. Classic neuropathological findings of GSS are its distinct large PrPSc amyloid plaques, called kuru plaques (Figure 9.3c), in association with pyramidal tract degeneration [48, 98, 99]. PrPSc amyloid plaques rarely are seen in other prion disorders, but their presence should make one consider PRNP testing. There is often gliosis but less vacuolation than classic sCJD [41]. GSS symptoms usually begin in the second to eighth decades (typically 20s–60s) with a mean duration of about 5 years [102], although a 15-year-old with a progressive motor and cognitive disorder beginning at age 10 was recently found by whole exome sequencing to have a de novo GSS mutation. FFI is a rare disorder that usually begins with progressive, severe intractable insomnia that is present for several months before onset of other symptoms, such as dysautonomia, ataxia, or other motor symptoms, with cognitive problems appearing later in the course. Progressive insomnia is eventually associated with hallucinations. Onset usually occurs in the fifth and sixth decade with duration of around 12–18 months [48, 61]. Although brain MRI is usually normal, FDG-PET imaging reveals thalamic and cingulate hypometabolism, often even before disease onset. FFI is caused by a single PRNP point mutation, D178N, with codon 129 M on the same chromosome (cis) (patients with D178N-129V usually present with fCJD) [110]. The neuropathology of FFI is primarily characterized by thalamic gliosis and neuronal loss [48, 98]. Kuru Kuru (“to shake or tremble” in the Fore language) [111] was a form of PrD confined to the Fore ethnic group of Papua New Guinea and was transmitted through a practice in which deceased relatives were honored by ritualized cannibalism. The clinical presentation was of pure cerebellar ataxia (and relatively preserved cognition) and an illness duration of 6–36 months [112]. The practice of cannibalism stopped in the late 50s, and since then, the incidence of kuru decreased dramatically (from the more than 2700 cases identified between 1957 and 2004, only 11 occurred after 1996) [113]. The mean incubation period was estimated to be around 12 years, but with a wide range from 5 to 56 years (particularly longer in those heterozygous at codon 129) [113]. Iatrogenic Jakob–Creutzfeldt disease More than 400 cases of iCJD have been reported, either from the use of cadaveric-derived human pituitary hormones, dura mater grafts, and corneal transplants, reuse of EEG implanted depth electrodes, and other neurosurgical equipments [114, 115]. The number of iCJD cases has been decreasing over the past years, probably due to increased surveillance and use of effective decontamination measures, but continuing surveillance is still necessary [114, 116]. The first recognized human-to-human transmission of CJD was reported in 1974, when a 55-year-old woman developed CJD 18 months after having inadvertently received a corneal transplant from a donor with CJD [117]. A second case associated with corneal transplant was reported years later, with an incubation period of 30 years [118]. In 1977, a report described two cases of iCJD caused by contaminated implanted stereotactic EEG electrodes that had been previously used in a CJD patient (incubation time of 16 and 18 months) [119]. Other neurosurgical instruments have also been implicated in iCJD cases [112], thus reinforcing the need of preventive measures when dealing with prions (see decontamination measures in the following). Cadaveric human pituitary hormones (human growth hormones [hGH] and human pituitary gonadotropic [hPG] hormones) were used for medical treatment from the late 1950s until mid-1985, and more than 30 000 patients are thought to have received them. In 1985, a report first mentioned the association between CJD and hGH, leading to the suspension of human pituitary hormone. The cases of hGH iCJD (~200 worldwide) occurred mainly in France, the United Kingdom, and the United States, and the incubation period was calculated to be around 15 years (range 4–36 years) [114]. The clinical presentation usually was of pure cerebellar ataxia, with dementia occurring only late (if at all) in the disease progression [112]. As in other forms of iCJD, PRNP codon 129 homozygosity is a risk factor of hGH iCJD [120]. hGH iCJD risk varies from country to country; in the United States, no cases were reported from individuals that received hormones after 1977 (when purification methods were changed), whereas in France there was a strong clustering of cases that received hormone between 1982 and 1985 [121, 122]. In the early 1990s, four cases of iCJD associated with hPG were identified in Australia, in women who had received the hormone in the 1970s as an infertility treatment [114, 121, 123]. No cases of hPG iCJD have been reported in other countries. The last major group of iCJD are those associated with cadaveric dura mater grafts, which was first recognized in 1987 [124]. Around 200 cases have been reported worldwide, and more than 60% occurred in Japan (but also in France, Spain, Germany, United Kingdom, and other countries) [114]. Lyodura© was the brand implicated in more than 90% of the cases, and no iCJD cases have been reported in patients who received the first dural graft in 1993 [112, 125]. The incubation period was calculated in the Japanese sample to range from 1.2 to 24.8 years, and the mean age of onset of symptoms was 55 years (range 15–80) [126]. Homozygosity at codon 129 (particularly M) is also a risk factor for this form of iCJD [114, 126]. Variant Jakob–Creutzfeldt disease vCJD was first recognized in 1995 in the United Kingdom [127] and soon received worldwide attention for its association with BSE, or mad cow disease. As of June 2014, approximately 225 vCJD cases have been identified, most in the United Kingdom and France [128]. BSE is the only non-hPrD currently believed to be transmissible to humans, and it is thought that BSE occurred from the practice of feeding scrapie-infected sheep products to cattle. More than 180 000 cattle suffered from BSE, the vast majority in the United Kingdom [129]. Although the incidence of BSE has dramatically declined since 1992, a few isolated cases have still been reported over the past few years [129]. But how was BSE diseased cattle’s meat consumption associated with vCJD? Since the first report of vCJD [127], a possible association between with BSE was raised due to the epidemiological temporal relationship between the two diseases. Compelling evidence soon came from experimental studies, in which similarities were found between BSE and vCJD PrP strains in mouse transmission studies, leading to the conclusion that they were caused by the same agent [130, 131]. It was later found out that vCJD could not only be acquired through contaminated beef consumption but also from blood product transfusion and there have been five cases reported so far with this association [132–136]. Four of those patients acquired vCJD from nonleukodepleted blood transfusions received before 1999, and one was a hemophiliac patient who received factor VIII in the 1990s from a contaminated batch. The incubation period ranged from 6 to 9 years in the three 129MM patients that died with definite vCJD. The other two patients (one acquired via blood transfusion, the other via factor VIII) were 129MV and died of nonneurological causes but had positive prion testing in their lymphoreticular system; it is not known whether they ever would have developed neurological PrD, although they were likely carriers. Because of this, additional measures were taken to prevent transmission of vCJD through blood products. Aside from donor selection and efforts toward developing methods to detect PrP in the blood, one of the main measures taken was universal leukoreduction of donated blood (which is being done since 1999 in the United Kingdom and later in the rest of Europe) [129]. In one study with hamsters, leukoreduction was shown to reduce TSE infectivity by 42% (comparing to whole blood) [137]. It is important to note that although transmission through blood products has been reported in vCJD, there are no known cases to date of transmission from sCJD patients through blood transfusion [138]. Codon 129 polymorphism is an important susceptibility factor for the development of vCJD, and almost every case reported so far has been 129MM (except for one symptomatic probable vCJD and the two presymptomatic blood product transfusion cases mentioned above) [139]. The clinical presentation of vCJD is different from sCJD in several ways. Patients with vCJD are usually younger, with a median age of onset around 27 (range 12–74) [140, 141]. The vast majority of cases occurred in persons younger than age 50; and among the patients in the United Kingdom, 12% died at age 20 or younger [142]. The mean disease duration is longer, about 14.5 months (vs. ~7 months for sCJD). Although psychiatric symptoms often occur early in sCJD [51], prominent psychiatric symptoms are often the initial symptoms in vCJD for several months (typically more than six) before obvious neurologic symptoms begin. The EEG only rarely shows the classic PSWCs and, if so, then only at the end stage of disease [143]. Brain MRI usually shows the “pulvinar sign,” in which the pulvinar (posterior thalamus) is brighter than the anterior putamen on T2-weighted (and probably also on DWI) MRI (and was found in more than 85% of cases in the first exam) (Figure 9.2d) [144]; this finding is very rare in other hPrDs [145, 146]. Posterior thalamic hyperintensities have been reported in gCJD (in E200K mutations) and in sCJD, but in those cases, the basal ganglia are usually brighter than the posterior thalamus [147]. Diagnostic criteria for probable vCJD are shown in Table 9.3 and have reported sensitivity of 83% and specificity of 100% [140]. Table 9.3 Diagnostic criteria for vCJD. Source: Modified with permission from Heath et al. [140]. * Spongiform change and extensive prion protein deposition with florid plaques throughout the cerebrum and cerebellum. † Depression, anxiety, apathy, withdrawal, and delusions. ‡ Includes frank pain and/or dysesthesias. § The typical appearance of the EEG in sporadic CJD consists of generalized triphasic periodic complexes at approximately 1 per second. These may occasionally be seen in the late stages of vCJD. ¶ Tonsil biopsy is not recommended routinely nor in cases with EEG appearances typical of sporadic CJD but may be useful in suspect cases in which the clinical features are compatible with vCJD and MRI that does not show bilateral pulvinar high signal. EEG, electroencephalography; MRI, magnetic resonance imaging; TSE, transmissible spongiform encephalopathy; vCJD, variant Jakob–Creutzfeldt disease. Definitive diagnosis of vCJD is based on neuropathologic evidence of the variant form of PrPSc in brain biopsy or autopsy. Because vCJD is acquired peripherally, PrPSc can be found in the lymphoreticular system, including tonsillar tissue [148]. Brain pathology of vCJD shows abundant PrPSc deposition, in particular multiple fibrillary PrP plaques surrounded by a halo of spongiform vacuoles (“florid” plaques) and other PrP plaques and deposits, especially prominent in the cerebellar molecular layer (Figure 9.3d) [98, 149]. From the blood product vCJD cases, it is now known that transmission from asymptomatic blood donors may occur years before the onset of symptoms, and so there is currently concern over the possibility of a second wave of vCJD in the future [150]. Exposed individuals with MV or VV genotype at codon 129 of the PRNP gene may also have longer incubation times, which could potentially increase the chance of a second wave. In an initial study in the United Kingdom, vCJD prions were found by immunostaining in 3 of 11 246 (~1 in 4000) appendix samples collected from 1995 to 2000 [151]. A more recent, larger study found that the rate of vCJD-affected appendices was 16 of 32 441 (~1 in 2000), about double the earlier study, suggesting an overall prevalence of 493 per million population [152]. Because the samples were anonymized, it is only possible to assume that there are asymptomatic persons in the UK population with vCJD prions in their lymphoreticular system (subclinically infected) who are at a minimum carriers of the disease. It is not clear what their risk is of developing symptomatic vCJD and/or transmitting it to others through medical/surgical procedures or blood products. In spite of all active efforts, there are no currently available drugs to change disease progression in hPrD, and so, symptomatic treatment is the only available option. Past efforts included quinacrine, an antimalarial drug, the antibiotic doxycycline, and the analgesic flupirtine, none of which recently have been shown to be effective in halting the progression of CJD [153–156]. Intraventricular pentosan polysulfate, an anticoagulant, was used in a few cases in the United Kingdom and in Japan, but its benefit is still uncertain. At best, it might prolong survival, but not affect disease progression and disability [157–160]. Symptomatic treatment may include the empirical use of SSRIs to treat depression and agitation, atypical antipsychotics (particularly quetiapine) to treat agitation and psychoses, and clonazepam to treat severe myoclonus or agitation. Two other management points are fundamental. As mentioned before, a significant percentage of gPrD have no evident positive family history (and sporadic PrD and gPrD may be clinically indistinguishable), and so genetic testing should be considered for every PrD patient (genetic counseling is indispensable prior to testing for PRNP mutations). Also, family and caregiver education is paramount in the disease process. In some countries around the world, there are organizations (such as the CJD Foundation in the United States; www.cjdfoundation.org) specialized in providing the necessary information on the care of patients with PrD [161]. More on family and caregiver issues will be discussed in Chapter 15. Decontamination of prions requires methods that will denature proteins, as prions resist normal inactivation methods used to kill viruses and bacteria. Typical methods for reducing the load of or inactivating prions include very high temperatures for prolonged periods, autoclaving at higher than normal temperatures, pressure, and time and with or without denaturing agents (many of which are caustic). Those measures often damage medical equipment and instrumentation [162]. WHO guidelines state that the preferred method is steam sterilization for at least 30 min at 132°C in a gravity displacement sterilizer. If a prevacuum sterilizer is used, they note 18 min at 134°C also is effective. Another option is 1M sodium hydroxide or 2% sodium hypochlorite for 1 h with 134°C autoclaving for at least 18 min. Nonfragile items may be immersed in 1N sodium hydroxide, a caustic solution, for 1 h at room temperature and then steam sterilized for 30 min at a temperature of 121°C [116, 163]. Due to the risk of transmission to subsequent patients, when feasible, many hospitals dispose of neurosurgical and other surgical equipment potentially exposed to prions by incineration. Our own medical center has developed its own policies and procedures for patients with suspected or confirmed hPrD, which are in some ways stricter than WHO guidelines, in part due to the large number of patients with PrD assessed at UCSF (http://infectioncontrol.ucsfmedicalcenter.org/sites/infectioncontrol.ucsfmedicalcenter.org/files/Sec%204.2%20Human%20Prion%20Policy.pdf). Aside from prion decontamination, additional preventive measures have been taken (some of which have been commented above) to avoid further iCJD cases. Known pathogenic PRNP mutation carriers are asked not to donate blood, even though the actual risk of transmissibility in humans is unknown [164]. Patients who received potentially contaminated human pituitary hormones were likewise advised in some countries [123, 165]. Other issues involving those at-risk individuals are still unknown (such as the necessary precautions needed to avoid transmission after a general surgery), pointing out the need for further research.
Prion diseases and rapidly progressive dementias
Rapidly progressive dementias
Prion diseases
Introduction
Pathophysiology
Prions
Genetics
Sporadic Jakob–Creutzfeldt disease
Clinical features
Diagnostic criteria
WHO 1998 revised criteria [72]
UCSF 2007 criteria [73]
European criteria 2009* [188]
MRI definitely CJD
DWI > FLAIR hyperintensities in:
MRI probably CJD
MRI probably not CJD
MRI definitely not CJD
Other MRI Issues
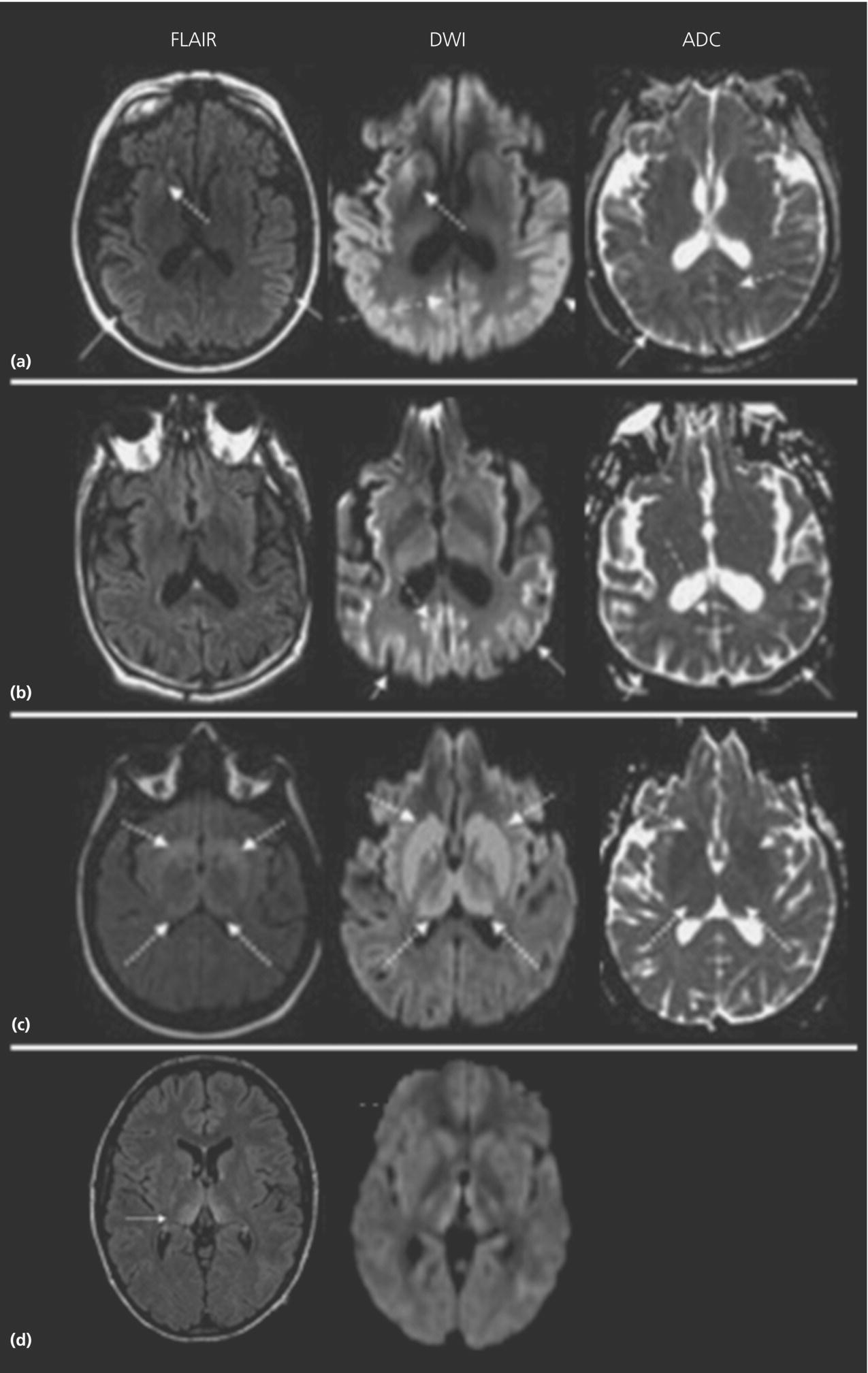
Neuroimaging
Laboratory and EEG findings
Blood tests
Cerebrospinal fluid
Electroencephalogram
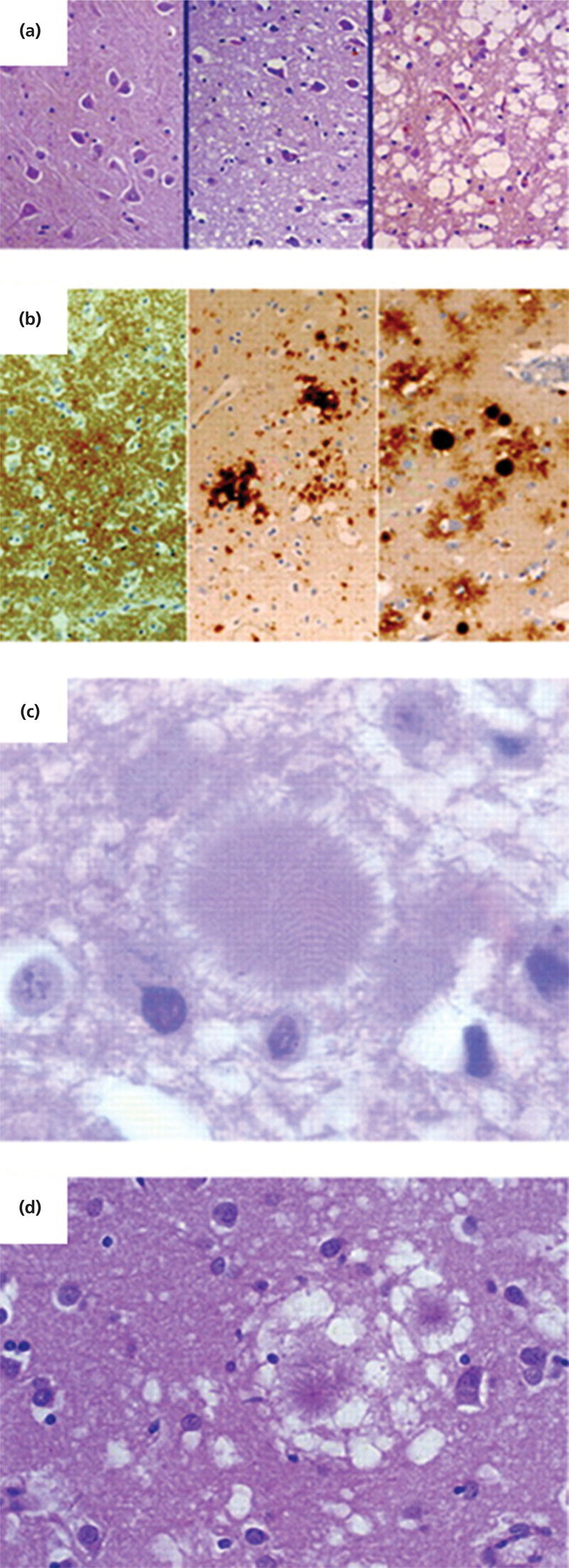
Neuropathology
Genetic prion disease
Introduction to gPrD
Genetic Jakob–Creutzfeldt disease
GSS
FFI
Acquired prion disease
Definite: IA and neuropathological confirmation of vCJD*
Probable: I and 4/5 of II and IIIA and IIIB or I and IVA.
Possible: I and 4/5 of II and IIIA
Treatment and management
Prion decontamination and preventive measures
Differential diagnosis of rapidly progressive dementias
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


