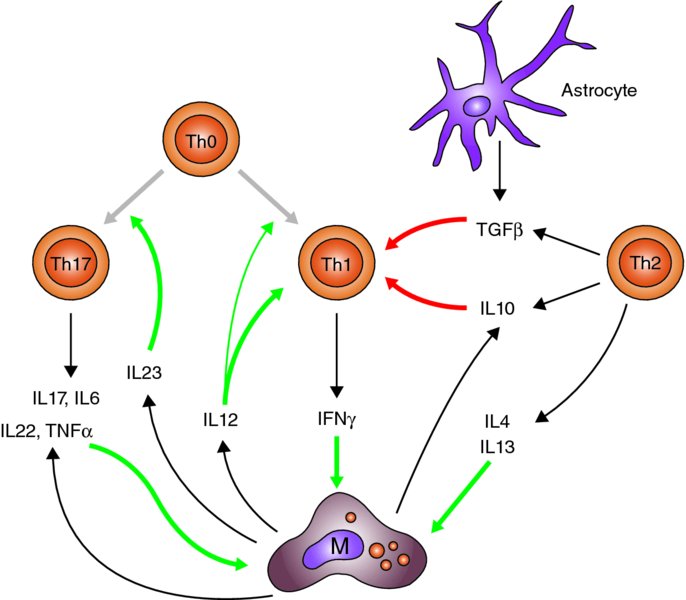
3 David Male Faculty of Science, The Open University, Milton Keynes, UK The adaptive immune response consists of two major lymphocyte populations, T and B lymphocytes. Both cell types have antigen-specific receptors on their cell surface: the T cell receptor (TcR) and the B cell receptor (BcR). Naïve and memory T cells are present within the circulation and respond differently to antigens presented in association with major histocompatibility complex (MHC) molecules on antigen-presenting cells (APCs). In addition to interaction between the TcR and MHC peptide on the APC surface, costimulatory molecules (CD80 and CD86) are required to promote T cell activation. T cells can be broadly divided into CD4 T helper cells (Th) which interact with MHC class II (MHC-II) molecules, and cytotoxic T cells expressing CD8, which interact with MHC class I (MHC-I) molecules on APCs. Antigen presentation takes place in six stages: If any of these functions are missing, then the APC will not fully activate the T cell. As a minimum, the APC must complete stages 1–5; lack of costimulation will result in T cell anergy. It is only if the T cell completes all stages that it will then undergo cell division. In practice, lymphocyte division does not occur in the central nervous system (CNS) to any significant extent, either because the threshold for T cell division is not reached, or because there are additional controls limiting lymphocyte proliferation. However if T cell activation occurs, then the release of inflammatory cytokines (interferon gamma (IFNγ), lymphotoxin etc.) can activate immune effector functions with the capacity to produce local damage within the CNS. The overall effect of these functions is not only to constrain immune reactions in the CNS but also, when they do occur, to polarize them towards Th1-type immune responses or in some cases Th17-type responses (see Figure 3.1). This polarization of response is seen both in the subsets of lymphocytes and phagocytes that accumulate, and in the way that cells become activated once they have entered the CNS. Figure 3.1 Cytokine networks in T cell activation. T helper cells (Th0s) can differentiate into Th17 cells or Th1 cells in response to IL23 or IL12, respectively. Th17 cells release cytokines, including TNFα, which promote macrophage activation. IFNγ released by Th1 cells also activates macrophages and promotes antigen presentation. The action of Th1 cells is counteracted by cytokines released from astrocytes, Th2 cells and in some cases microglia. M = Mononuclear phagocytes, including macrophages and activated microglia; white arrows = differentiation; black arrows = production; red arrows = suppression; green arrows = activation. The profile of leukocyte populations seen in neuroinflammation is highly selective. It is worth noting that neutrophils, which are the most abundant leukocytes in the blood (70%), are rarely seen in the CNS, and if so, only in stroke and the most severe cases of acute encephalitis. This is exactly the opposite of what is seen in inflammation in most other tissues, and in bacterial meningitis, where neutrophils predominate. This observation alone indicates that distinctive mechanisms control leukocyte migration into the CNS. The major leukocytes seen in CNS inflammation are lymphocytes and mononuclear phagocytes (derived from blood monocytes), a pattern more typical of sites of chronic inflammation. Initial observations of multiple sclerosis (MS) post-mortem tissue showed that both CD4+ and CD8+ T cells were present in chronic active inflammatory lesions, with CD4+ cells extending out beyond the lesion and with a relatively high proportion of CD8+ cells at the active edges of the plaques (Traugott et al., 1983). These plaques were often examples of the late stages of a chronic disease, and more active plaques contained higher proportions of CD4+T cells. Studies of experimental autoimmune encephalomyelitis (EAE) in rodents confirmed the importance of CD4+ T cells in the development of acute neuroinflammation. Not only were CD4+ helper T cells prevalent in the spinal cord during the acute phase of disease, but also the disease could be passively transferred with antigen-activated CD4+ T cells. Moreover, disease development could be blocked with antibodies to CD4 or MHC-II molecules, which interfere with antigen presentation to CD4+ T cells. Although this may show that CD4+ T cells play a dominant role in disease induction, Sun et al. (2001) also describe the encephalitogenic potential of CD8+ T cells in mice. Interestingly, however, in passive transfer EAE, the great majority of the T cells found in the affected areas were not donor cells, but host cells and therefore probably not specific for any CNS antigen – although recognition of CNS antigens is required for the development of neuroinflammation, lymphocyte migration itself is not antigen specific. The process explained in this chapter does, however, favour the accumulation of T cells that have been recently activated by specific antigens in the periphery of the body. The recognition of subsets of CD4+T cells that secrete different cytokine profiles led to a more thorough investigation of the cell types responsible for neuroinflammation (see Table 3.1). Th1 cells characterized by the secretion of IFNγ respond to antigen presented by mononuclear phagocytes, and are effective at inducing expression of MHC molecules and activating macrophages. Th2 cells secrete IL4, IL10 and IL13 and are more effective at interacting with B cells as APCs, and their cytokines promote B cell development and antibody secretion. The finding of IFNγ in lesions in MS in association with activated macrophages and microglia strongly implied that Th1 cells were primarily responsible for development of lesions. IL12 and IL18 secreted by mononuclear phagocytes also contribute to polarizing the T cell phenotype to Th1 in MS, with IL12 promoting Th1 cell differentiation and IFNγ secretion and IL18 promoting tumour necrosis factor alpha (TNFα) production. This pattern of Th1 cytokine production is found in active MS lesions. Table 3.1 T cell phenotypes, the cytokines which induce their polarization and the effector cytokines produced by each subset GM-CSF = granulocyte–macrophage colony-stimulating factor; TGFβ = transforming growth factor beta. The initial observations in EAE tended to support this view. It is possible to generate Th1-type or Th2-type antigen-specific T cells in vitro by treating the cells with different combinations of cytokines and growth factors. In this case, Th1 cells secreting IFNγ, IL2 and TNFα could transfer EAE, whereas Th2 cells that had an identical antigen specificity but secreted IL4, IL5, IL10 and IL13 did not. The theory was further supported by observations that strains of animals that produce strong antibody responses against CNS antigens often do not develop severe EAE. A major problem with the hypothesis that Th1 cells are responsible for neuroinflammation arose from the finding that some strains of mice with IFNγ receptor deleted developed more serious disease than wild-type animals, and it was possible to induce EAE in TNF knockouts. Emphasis then switched to the investigation of another subclass of CD4+ T cells, Th17 cells, characterized by the production of IL17, IL6, IL22 and TNF. This population of cells is induced and maintained by IL23, produced by inflammatory macrophages and dendritic cells. It was found that mice deficient in IL23 were resistant to the development of EAE, whereas deletion of IL12 did not prevent disease (Cua et al., 2003). Moreover, treatment with anti-IL23 antibody could alleviate EAE in one model, whereas anti-IFNγ did not (Chen et al., 2006). In EAE, Th17 cells promote disease by acting at the choroid plexus, a site which has quite different properties and endothelium compared to the normal CNS parenchyma (Reboldi et al., 2009). Interpreting studies from knockout animals does present some difficulties, because deficiency in one immunological pathway can lead to compensatory activity in others. Nevertheless, the work in EAE lead to a re-examination of the T cell subsets in MS, with the findings that Th17 cells were present and associated with active disease (Tzartos et al., 2008) and that activated microglia secrete IL23 (Li et al., 2007). Zielinski et al. (2012) very recently described two types of Th17 cells with different inflammatory properties, producing either IFNγ or IL10, the former representing pathogenic Th17 cells and the latter non-pathogenic cells. IL1β induced Th17 cells producing IFNγ while suppressing IL10 production, and they suggest that IL1β rather than IL23 promotes polarization of highly inflammatory Th1 and Th17 responses in human CNS. A summary of the interactions between the CD4+ lymphocyte subsets and the role of the different cytokines is shown in Figure 3.1. In viewing schemes of this type, one should be aware that the cytokine production profiles of different cells are variable and may have pleiotropic effects. For example, microglia can secrete IL12 (Th1-type response), IL10 (Th2-type response) and IL23 (Th17-type response). The role of CD8+ T cells in neuroinflammation has been partly overshadowed by work on CD4+ T cells, even though they sometimes are the predominant cell type. Indeed, some studies have demonstrated the induction of variants of EAE, using CD8+ T cell clones; however, these models are the exception to the general rule that activated CD4+ T cells are required to induce EAE (Mars et al., 2011). In some cases, CD8+ T cell clones are expanded in the cerebrospinal fluid (CSF) of MS patients, and such clones can persist for many years (Skulina et al., 2004). It has been argued that whereas CD4+T cells and macrophages cause demyelination and damage to oligodendrocytes, CD8+ cells could contribute to the subsequent damage to exposed axons or neurites (McFarland and Martin, 2007). Tregs, identified by the transcription factor Foxp3 and high levels of the IL2 receptor CD25, are important in controlling tissue-specific autoimmunity and inflammation, particularly in the gut. They are thought to act by the release of anti-inflammatory cytokines including IL10, IL35 and TGFβ (Korn et al., 2007). More recently, a population of CD8+ Tregs producing TGFβ and (surprisingly) IFNγ have been shown to suppress EAE induced by myelin oligodendrocyte glycoprotein (MOG) in SJL mice (Chen et al., 2009). These observations led to investigations of Tregs in MS. The incidence of Tregs in the blood and the CNS of MS patients appears to be similar to that found in controls, but there is some evidence that the functional activity of Tregs is lower in individuals with relapsing and remitting MS than in those with the progressive disease (Venken et al., 2006). Oligoclonal antibodies (i.e. antibodies produced by a small number of B cell clones) are present in the CSF of patients in most infectious diseases of the CNS as well as in MS. Since serum antibodies are effectively excluded by the blood–brain barrier (BBB), this fact alone indicates that some B cells must be present in the brain. The clones represent a small subset of those present in the blood (Owens et al., 2001); however, these clones can be stable over many years, which is longer than the normal lifespan of a plasma cell. This implies that the CNS environment, in some way, allows clones of B lymphocytes to persist. One hypothesis that could explain the persistence of B cells is infection with Epstein–Barr virus (EBV), a recent candidate virus in MS. Although this is an attractive hypothesis, there is still debate regarding the presence of EBV in MS tissues (Lassmann et al., 2011). B cells and plasma cells were first observed in the Virchow–Robin spaces around larger blood vessels in MS, which explains why they could be detected in the CSF. Although in infection the antibody is usually specific to the pathogen, the antibody targets in MS and other inflammatory conditions were often not identifiable. This led to discussion as to whether these antibodies were merely an epiphenomenon (i.e. random local antibody production, caused by a few persistent B cells that had entered the perivascular space). This has never been a wholly satisfactory explanation, because antigen is normally required to drive the differentiation of B cells to plasma cells and to maintain an antibody response over time. Moreover, antibody-producing cells do occur in early MS lesions (Henderson et al., 2009), and B cell depletion can alleviate relapses in MS (Hauser et al., 2008). In EAE it appears that small amounts of antibody are important in targeting macrophages against oligodendrocytes, regardless of the immunological target of the T cells. For example, EAE can be induced by an immune response against astrocyte S100B protein or neuronal antigens such as neurofilament light chain (NF-L) (Huizinga et al., 2007). However, myelin damage is clearly due to antibodies to oligodendrocytes, especially MOG (Smith et al., 2005). Consequently, antibody may be required to target the immune reaction even if neuroinflammation is primarily driven by Th1 and Th17 cells. The level of leukocyte traffic through the CNS is normally low in the absence of neuroinflammation; hence, there are relatively few lymphocytes present in the normal CNS. Lymphocyte migration into the CNS involves binding to the endothelium following selectin-mediated rolling and subsequent chemokine-mediated integrin activation, leading to firm adhesion and transmigration across the BBB. Within the inflamed CNS, chemokines direct lymphocytes, expressing the appropriate chemokine receptors to sites of inflammation. Lymphocyte migration into the CNS takes place primarily across post-capillary venules, and this is reflected in the location of lymphocytes in the CNS, in perivascular cuffs as seen in MS pathology. The cuff is formed by tightly packed lymphocytes and monocytes in the space between the endothelium and the astrocyte foot-processes, effectively between the double basal lamina produced by the two cell types. Leukocytes are then able to break through the glia limitans barrier following secretion of matrix metalloproteinases (Agrawal et al., 2013) to enter the brain parenchyma. Lymphocyte migration into the CNS is controlled, as in other tissues, by chemokines and adhesion molecules expressed on the luminal side of the brain endothelium (Male, 2012). These controls are tissue specific, so that the pattern of lymphocyte migration into the CNS, and hence the types of immune reaction that can occur, is constrained by these controls (Engelhardt and Ransohoff, 2005; Man et al., 2007). The BBB prevents the diffusion of chemokines from the CNS to the blood side of the endothelium, via the paracellular route. Additionally, resting brain endothelium has comparatively few caveolae and other transport vesicles, which limits movement of chemokines by transcytosis. This means that chemokines produced by the brain endothelium make a particularly large contribution to the control of lymphocyte migration into the tissue, by comparison with endothelium in other tissues. Reboldi et al. (2009) demonstrated in EAE that the choroid plexus is the first port of entry of inflammatory T cells into the CNS. Specifically, CCR6 expressing Th17 cells migrate in response to the CCR6 ligand, CCL20, expressed by the epithelial cells of the choroid plexus (Figure 3.2). Sallusto et al.
Adaptive Immune Responses in the CNS
Introduction to concepts of adaptive immunity
Leukocyte populations in the CNS
T cell populations
T lymphocyte subsets
Th1, Th2 and Th17 cells
T cell phenotypes
Polarizing cytokines
Effector molecules
Function
Th1
IL12, IFNγ
IFNγ
Macrophage activation; induction of MHC II
Th2
IL4
IL4, IL5 and IL13
Interact with B cells as APCs; Th2 cytokines promote B cell development and antibody secretion
Th17
IL6, IL1β, IL23 and TGFβ
IL17, IL22 and GM-CSF
Promotes acute inflammation
Treg
IL10, IL35 and TGFβ
Suppresses/regulates other T cell subsets
Cytotoxic T lymphocytes
Regulatory T cells (Tregs)
B cells
Mediators of the adaptive immune response
Control of lymphocyte migration into the CNS
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel







Full access? Get Clinical Tree


