Plasmid
Ratio
Amount per plate (μg)
Amount per 30 dishes transfection (μg)
Helper plasmid
2
26
780
Packaging plasmid
1
13
390
cis plasmid pCMV-GFP
1
13
390
Transfection by calcium phosphate precipitation
Calcium phosphate transfection, which was initially described more than 50 years ago, has been refined and improved to result in a standard protocol which is widely used in the production of recombinant viral vectors. This protocol works best in highly transformed adherent cells, obtaining transfection efficiencies from 20 to 100 %, depending on the cell line (HeLa, U2OS, SAOS2, AdAH, NPC-KT). HEK293T cells, a popular producer cell line for AAV vectors, allow for transfection efficiency of up to 90 %.
The technique relies on precipitates of plasmid DNA formed by its interaction with calcium ions. It is inexpensive and technically easy to perform. Plasmid DNA is mixed in a solution of calcium chloride, and then is added to a phosphate-buffered solution. Over a period of 10–20 min, a fine precipitate carrying a net positive charge forms in the solution and becomes associated with the plasmid DNA. This suspension is then added directly to the cells in culture.
The mechanism by which this complex enters cells is not known in detail, although it has been suggested that it is first drawn into acidifying endosomes before DNA is transferred to the nucleus. The transfer of DNA across the nuclear membrane must occur during mitosis. Therefore, expression of the gene of interest will not occur until the cells have passed through this phase.
Although this technique has minimal cellular toxicity and is both simple and inexpensive, the relatively low level of transgene expression using some cell lines prompted development of other techniques.
1.
Thaw plasmids to ensure complete DNA resuspension. Prewarm the following solutions to 37 °C:
2.5 M CaCl2
2× HBS, pH 7.05 (the pH is critically important)
Complete DMEM media with 5 % FBS, Pen-Strep, and l-glutamine.
2.5 M CaCl 2 : 36.75 g CaCl2 · 2H2O (Sigma C7902) in 100 ml dH2O, sterilized by filtration through a 0.22 μm filter. Store at −20 °C.
2× HBS: 280 mM NaCl, 1.5 mM Na2HPO4, 50 mM HEPES. Adjust to pH 7.05 with NaOH and sterilize by filtration. Store at −20 °C.
(Note: The pH of HBS is extremely important as even relatively small deviations can negatively affect transfection efficiency)
2.
Prepare three 50 ml tubes (Tube 1, each will serve for transfection of 10 dishes) and add 1.25 ml CaCl2 to each tube plus the calculated amounts for each of the three plasmids: helper plasmid, packaging plasmid, cis plasmid, e.g., pCMV-GFP. Make up to 12.5 ml with sterile water.
Prepare three more tubes (Tube 2) and add 12.5 ml of 2 × HBS.
Place Tube 1 on a holder and gently bubble it slowly by expelling air using pipette and electronic pipette aid. Then add the content of Tube 2 drop-wise into Tube 1 using a 1 ml pipette. Once all solution has been added, allow the mixed solution to settle for 10 min at 37 °C. The mixture should appear slightly opaque and milky.
3.
To a tissue culture flask, add 200 ml of prewarmed serum-free media and then add 25 ml transfection mixture.
Aspirate the medium from the 30 dishes and add 20 ml of the medium with DNA precipitates into each dish. Mix the suspension as you go along to prevent the DNA precipitates from dropping to the bottom of the container. Be careful not to displace the cells from the dishes.
4.
Transfer the dishes to the incubator and leave for 48 h.
3.6 Preparation of Cell Lysate for Ultracentrifugation in Iodixanol Gradient
1.
48 h after transfection aspirate all but 3–5 ml medium from each dish, scrape the cells off using a cell scraper, and transfer the media/cells into 50 ml Falcon tubes. Wash the dishes with 10–15 ml complete DMEM or PBS (starting from dish No. 1 and then transferring the medium to the next dish till all dishes are washed) to harvest all the cells.
2.
Centrifuge at 1,200 × g for 10 min at room temperature and aspirate supernatant.
3.
Resuspend cell pellets in 15 ml Lysis buffer (150 mM NaCl, 50 mM Tris–HCl, pH 8.5) per 10 dishes.
4.
Freeze/thaw three times by alternate immersion in dry ice/ethanol bath and 37 °C water bath.
5.
Add Benzonase nuclease to the lysate to a final concentration of 50 U/ml (750 U/15 ml). Incubate at 37 °C for 30–60 min.
6.
Centrifuge at 4,000 × g for 20 min and transfer the vector-containing supernatant to a clean tube. It may be used immediately or stored at 4 °C for several hours while the iodixanol gradients are prepared.
7.
Proceed to Sect. 3.8 for description of the tube loading, ultracentrifugation, and subsequent steps. Note that a single Quick-Seal 39 ml centrifugation tube (Beckman Coulter #344326) can accommodate up to 15 ml of lysate, and the lysate from no more than 15 dishes should be loaded into a single tube. More concentrated material would compromise the resolution and the purity of the vector.
3.7 Polyethyenimine (PEI) Transfection
As an alternative to calcium phosphate, polyethyenimine (PEI) can be used to condense DNA and act as a carrier into targeted cells. PEI condenses DNA into positively charged particles, which bind to anionic cell surface residues and are brought into the cell via endocytosis. Once inside the cell its relatively strong positive charge combined with the acidification of the endosome results in an influx of counter-ions and a lowering of the osmotic pressure. Osmotic swelling occurs and the resulting osmotic gradient across the endosome’s membrane leads to an influx of water, bursting the endosome and allowing the PEI and bound DNA (polyplex) to escape to the cytoplasm. If the polyplex unpacks, then the DNA is free to diffuse to the nucleus. Further details of the DNA’s transit to the nucleus are not well established, but the extensive use has shown that PEI is an efficient transfection reagent, yielding transfection efficiencies and viral titers similar to the calcium phosphate precipitation method.
High charge density in physiological media and high buffer capacity in weakly acidic media account for PEI’s efficiency. Though low molecular weight PEIs are less toxic, their gene transfection efficiency is much lower than high molecular analogs such as 25 kDa PEI. The gene transfer efficiency of PEI is attributed to its unique ability to overcome a specific barrier to gene transfer, lysosomal degradation. The trafficking and subsequent degradation of vectors inside lysosomes is one of the main cellular barriers to effective gene transfer.
The protocol for PEI transfection is quite similar to that for calcium phosphate transfection, as described above. PEI (25 kDa linear PEI, Polysciences, Inc., cat. No. 23966) is prepared as a stock solution at a concentration of 1 mg/ml in water and pH is adjusted to 7.1. PEI is added to water, slowly warmed to approximately 50 °C, and vortexed until completely dissolved (this can take many minutes of vortexing). Once fully dissolved, PEI can be sterilized by filtration through a 0.22 μm syringe filter, aliquoted, and stored at −20 °C.
1.
16–20 h before transfection, seed 30 × 15 cm dishes with HEK239T cells to approximately 70 % confluence. Culture the cells in DMEM supplemented with 10 % FBS, 2 mM l-glutamine, and 1 % pen/strep.
2.
2 h before transfection, replace the medium with 16 ml of serum-free DMEM (supplemented with antibiotics if required). Dishes should be transfected when they are approximately 80–85 % confluent.
3.
To six 50 ml Falcon tubes, aliquot 15.5 ml of serum-free DMEM. Of these six tubes, label three “DNA” and three “PEI.”
4.
As described in the calcium phosphate precipitation, calculate the volumes of plasmid DNA required to transfect 30 dishes. Combine these three plasmid stocks and mix well by pipetting.
5.
Transfer equal volume of this plasmid mixture into each of the tubes labeled “DNA” and mix with the serum-free DMEM by vortexing briefly. Each tube should contain approximately 520 μg of DNA.
6.
Add 1,560 μl of PEI solution (PEI:DNA = 3:1 ratio; 1 mg/ml) to each of the tubes labeled “PEI” and mix with the serum-free DMEM by vortexing briefly.
7.
Add plasmid DNA solution to PEI solution and immediately mix by vortexing for 10 s. After thorough mixing, incubate at room temperature for 15 min to allow complexes to form.
8.
Depending on the volume contributed by plasmid DNA solutions, each of the three tubes (DNA-PEI complexes in serum-free medium) will contain around 34 ml, sufficient for transfection of ten 15 cm dishes.
9.
Add 3.3 ml of transfection mixture to the medium of each dish, distribute well by gentle agitation, and return cells to the incubator.
10.
24–48 h after transfection, transfection efficiency may be monitored by flow cytometry or fluorescence microscopy if the construct contains a fluorescent transgene under a suitable promoter.
11.
Two days after transfection, gently add 10 ml of fresh serum-free medium to each dish.
(NB: Step 11 is optional; it helps to maintain cells in better condition and may therefore improve virus yield, but the increase in volume will lengthen the purification steps.)
12.
Five days after transfection, add Benzonase nuclease to each dish to the final concentration 25 U/ml. Incubate at 37 °C for a further 2 h.
(NB: Instead of step 12, Benzonase may be added directly to the pooled supernatant in step 14. Add Benzonase to a final concentration of 25 U/ml, incubate at 37 °C for 2 h, and then proceed to clarify the lysate by centrifugation as described in step 14.)
13.
To each dish, add NaCl solution (filtered through 0.22 μm Nalgene Thermo Scientific Rapid-Flow filter) to a final concentration of 500 mM and incubate for 2 h.
14.
Collected medium from all dishes is pooled to a final volume of approximately 1 l. Since it contains cell debris, it must be clarified by centrifugation (3,850 × g for 5 min, Sigma 3-16PK, rotor 11180), followed by filtering through a 0.22 μm vacuum filter (Thermo Scientific Nalgene Rapid-Flow filter unit). Filtered supernatant may be used immediately or stored at 4 °C overnight.
15.
Finally, concentrate the supernatant by approximately 75 times. In the absence of tangential flow filtration equipment, Amicon Ultra-15 Centrifugal 100K Filters (Millipore) can be applied. Supernatant is centrifuged at 3,800 × g at 4 °C until the volume is reduced to 14 ml. While not laborious, this step can take several hours. Keep the supernatant refrigerated at all times.
3.8 AAV Purification by Iodixanol Density Gradient
For the iodixanol gradient ultracentrifugation use Quick-Seal 39 ml tubes (Beckman Coulter #344326) by underlaying and displacing the less dense supernatant. The solutions must be added slowly in order to prevent mixing of the layers and bubbles introduction by inserting a 100 mm, 18 gauge blunt-end needle (Hamilton #7750-09) and filling the tube from the bottom upwards.
1.
Preparation of iodixanol solutions:
Percentage iodixanol | Iodixanol | 5 M NaCl | 5 × PBS-MK (see below) | H2O | Phenol Red |
15 % | 12.5 ml | 10 ml | 10 ml | 17.5 ml | – |
25 % | 20.8 ml | – | 10 ml | 19.2 ml | 100 μl |
40 % | 33.3 ml | – | 10 ml | 6.7 ml | – |
54 % | 45 ml | – | – | 5 ml | 100 μl |
2.
The iodixanol solutions are layered very carefully using disposable syringes in the following order:
(a)
Virus solution (13–14 ml), concentrated from 30 dishes.
(b)
4 ml of 15 % iodixanol/1 M NaCl in PBS-MK buffer (1× phosphate-buffered saline (PBS), 1 mm MgCl2, and 2.5 mM KCl).
(c)
9 ml of 25 % iodixanol in PBS-MK buffer containing Phenol Red (slightly pink).
(d)
9 ml of 40 % iodixanol in PBS-MK buffer.
(e)
5 ml of 54 % iodixanol containing Phenol Red (slightly yellow).
3.
The tube is then filled up to the bottom of the neck (as per manual description) with PBS using either a pipette or a syringe with a small gauge needle, avoiding creating bubbles.
4.
The tubes are then securely sealed using the recommended heat sealing device to prevent leakage during centrifugation (Fig. 1a).
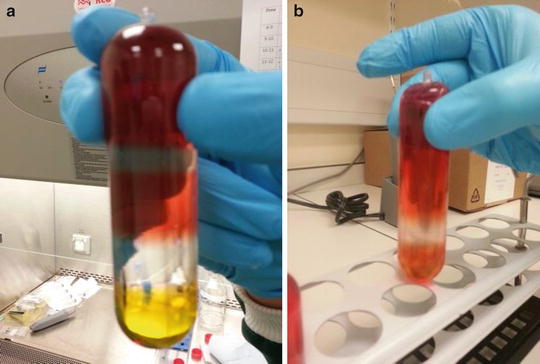
Fig. 1
AAV purification by iodixanol density gradient. Addition of increasing concentrations of iodixanol results in the formation of discrete layers prior to ultracentrifugation (a) with the least dense concentrated virus solution at the top of the tube. Following ultracentrifugation, viral particles are contained within the clear 40 % iodixanol layer (b) allowing for ease of extraction by fractionation
5.
The tubes are transferred to the Type 70Ti rotor (Beckman Coulter) and centrifuged at 69,000 rpm for 1 h 30 min at 18 °C using maximum acceleration but no braking on deceleration.
6.
After centrifugation the tubes are clamped in a retort stand and a 19 gauge needle is inserted into the top of the tube. The clear fraction of the 40 % layer now contains the virus (Fig. 1b). In order to extract the pure virus sample and to separate it from the debris and empty capsids, the best method to employ is fractionation. For this, a standard 19 gauge (or slightly thinner) syringe needle is inserted approximately 1 cm from the bottom of the tube. It should be pushed half way into the tube with the bevel pointing upwards. The placing of the needle should be such that it is towards the upper portion of the 60 % iodixanol layer. It is important to remember to puncture the top of the tube first so as to allow the liquid to flow. Placing the needle in this manner should cause slow leakage of the solution out of the tube in a drop-wise fashion. The drops are then collected in samples of approximately 250 μl in 1,500 μl Eppendorf tubes. The entire 40 % layer and the first portion of the 25 % layer are collected.
7.
This process is repeated for subsequent tubes and the collected fractions may be stored at 4 °C until further analysis provided it is carried out within a few days.
8.
4 μl of each fraction is diluted with water, combined with 4× reducing loading buffer, boiled and run on 10 % SDS-PAGE. Capsid proteins of AAV are visualized by SYPRO Ruby staining and fractions containing pure virus are pooled (Fig. 2). Only the fractions containing bands for the three capsid proteins are pooled as high-quality virus. Fractions with some higher molecular weight bands may also be pooled separately to be used in in vitro validation studies.
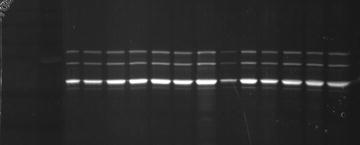
Fig. 2
Viral sample fractionation. Collected fractions were analyzed on 10 % polyacrylamide gel stained with SYPRO Ruby; fractions collected here were all pooled as high-quality virus as no contaminating bands were detected. These collected fractions were then taken for further concentration
3.9 Concentration and Desalting of AAV Preparations
1.
AAV is concentrated and desalted by centrifuging through a BIOMAX 100 Ultrafree 15 centrifugal filter device (Millipore UFV2BHK 10 or 40) or Amicon Ultra centrifugal filter device (Millipore UFC910008).
2.
Place the filter device in a 50 ml Falcon tube and add 15 ml of PBS-35 mM NaCl to the sample reservoir. Centrifuge at 3,000 × g for 3 min. Approx 500 μl of liquid should remain in the sample reservoir.
3.
Add the pooled viral samples from ultracentrifugation to the sample reservoir and fill up to 15 ml with PBS-35 mM NaCl and spin for 15–20 min. Check the speed of filtration after 3 min. The sample should be concentrated by about tenfold. There should be no more than 300–500 μl left in the sample reservoir. Spin for longer if the volume retained is higher than 500 μl.
4.
When the volume retained reaches ~500 μl, pipette the solution up and down onto the side of the filter device for about five times on each side. Be careful not to pierce/touch the filter.
5.
Add a further 15 ml of PBS-35 mM NaCl buffer and spin again. Increase spin time by approximately 10 min as the spin will take successively longer to reduce the volume of the sample. After 3–5 min check the filtration speed. Final volume should be under 700 μl (generally 200–500 μl).
6.
 Gene Therapy Approaches to Promoting Axonal Regeneration After Spinal Cord Injury
Gene Therapy Approaches to Promoting Axonal Regeneration After Spinal Cord Injury
 Gene Therapy Approaches Using Reproducible and Fully Penetrant Lentivirus-Mediated Endogenous Glioma Models
Gene Therapy Approaches Using Reproducible and Fully Penetrant Lentivirus-Mediated Endogenous Glioma Models
 Gene Therapy for Huntington’s Disease
Gene Therapy for Huntington’s Disease
 Gene Therapy for Epilepsies
Gene Therapy for Epilepsies
 Gene Therapy for Chronic Pain: How to Manipulate and Unravel Pain Control Circuits from the Brain?
Gene Therapy for Chronic Pain: How to Manipulate and Unravel Pain Control Circuits from the Brain?
 Gene Therapy for Parkinson’s Disease: AAV5-Mediated Delivery of Glial Cell Line-Derived Neurotrophic Factor (GDNF)
Gene Therapy for Parkinson’s Disease: AAV5-Mediated Delivery of Glial Cell Line-Derived Neurotrophic Factor (GDNF)




Repeat steps 4 and 5 until the original volume of the pooled fractions is exchanged at least ten times and until the desired volume of the vector in final formulation buffer is achieved.
Related posts:
Gene Therapy Approaches to Promoting Axonal Regeneration After Spinal Cord Injury
Gene Therapy Approaches Using Reproducible and Fully Penetrant Lentivirus-Mediated Endogenous Glioma Models
Gene Therapy for Huntington’s Disease
Gene Therapy for Epilepsies
Gene Therapy for Chronic Pain: How to Manipulate and Unravel Pain Control Circuits from the Brain?
Gene Therapy for Parkinson’s Disease: AAV5-Mediated Delivery of Glial Cell Line-Derived Neurotrophic Factor (GDNF)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
