Fig. 1
Basic vertebrate retinal structure image showing the various layers of the vertebrate retina with light passing through the ganglion cell layer (GCL), inner nuclear layer (INL) to the outer nuclear layer (ONL), and photoreceptor outer segments where light is absorbed
The clinical diagnosis of retinitis pigmentosa (RP) is the most common presentation of inherited retinal degeneration and affects approximately 1 in 4,000 people [4]. This clinical diagnosis encompasses a spectrum of genetic conditions with onset ranging from childhood to late adult life, usually featuring night blindness followed eventually by central visual loss, consistent usually with rod photoreceptor loss preceding that of cones. Inheritance may be autosomal dominant, autosomal recessive, X linked, or mitochondrial, and over 180 causative genes have so far been identified (https://sph.uth.tmc.edu/retnet/sum-dis.htm). Retinal features of loss of rod photoreceptors followed by cones lead to a secondary migration of the RPE into the retina giving a characteristic “bone spicule” appearance. Other retinal degenerations include cone–rod dystrophies (where cone loss precedes that of rods) such as in ABCA4 (ATP-binding cassette, subfamily A, member 4) mutations or nonprogressive retinal dysfunction syndromes such as achromatopsia where there is a loss of cone function without significant degeneration.
Gene therapy is a promising treatment for inherited retinal disease, and there are multiple approaches that need to be tailored according to the pathology of interest. In autosomal recessive conditions where a mutated gene results in a nonfunctional gene product, gene replacement therapy could potentially slow degeneration and restore function. In autosomal dominant conditions where an abnormal protein may cause toxicity, therapy may involve “knockdown” of the abnormal gene and expression of the correct allele or in some cases overexpression of the native protein may be adequate. In conditions where degeneration is advanced or the causative mutation is unknown, an optogenetic strategy (making non-photoreceptor retinal neurons light sensitive) may be more appropriate to restore vision. This involves using gene therapy to introduce a light-sensitive protein such as channelrhodospin-2 into residual retinal cells [5], making them intrinsically photosensitive in the absence of photoreceptors. Another nonspecific approach is to use gene therapy to induce expression of a neuroprotective protein such as ciliary neurotrophic factor (CNTF) [6] to reduce or arrest loss of cells in retinal degenerations.
A number of animal models for retinal diseases are available for the testing of gene therapy vectors, facilitating translation of research into the clinical domain. The condition in which the greatest developments in retinal gene therapy have been made is Leber’s congenital amaurosis (LCA) which results from mutations in RPE65 (retinal pigment epithelium-specific 65 kDa protein), a rare severe retinal dystrophy with autosomal recessive inheritance that manifests with poor vision at birth or within the first few years of life. Despite the loss in retinal function, the structure of the retina remains largely intact, making this condition an ideal target for gene therapy since potential for functional improvement remains. There are three murine models of RPE65 deficiency: an Rpe65 −/− knockout mouse [7], a naturally occurring Rpe65 mutation in the rd12 mouse [8], and an Rpe65 R91W/R91W knockin mouse which shows a partial expression of a mutant RPE65 protein [9]. Gene replacement therapy has been performed in the first two of these models using an adeno-associated viral (AAV) vector and in the third model using a lentiviral vector. Subretinal delivery of RPE65 to the Rpe65 −/− knockout mouse using AAV2 under the control of a ubiquitous chicken β-actin (CBA) promoter resulted in an RPE65 expression in the RPE and electroretinogram (ERG) improvement in four out of ten eyes [10]. AAV5 [11] and AAV2 [12] vectors have been assessed in the rd12 mouse both of which showed improvement in ERG function. In addition, a canine model of RPE65 mutation exists, the Briard dog, which has also been treated with AAV2 expressing RPE65 under a CBA promoter, with restoration of visual function observed [13]. These and other animal studies provided proof of principle that AAV retinal gene therapy for RPE65 could be administered safely and give efficacious results, paving the way for translation to patients.
In 2008, results from three human clinical trials of ocular gene therapy for RPE65 mutations causing LCA were published [14–16]. These initial reports from three independent groups at University College London, the Children’s Hospital of Philadelphia (CHOP), and the University of Florida/University of Pennsylvania each reported outcomes from three patients. All had a subretinal injection of an AAV2 vector containing RPE65 cDNA to target the RPE, although there were some differences between studies such as vector design, injection volume, and use of steroids. In all of the studies, no significant immune response was found to the vector or transgene product. Three patients showed improvement in visual acuity [15], and increased light sensitivity was found in seven out of nine patients.
Further publications from these centers along with a fourth center in Israel [17] have now detailed a total of 31 patients of age range 8–30 that have been treated. Jacobson et al. [18] provide up to 3 years follow-up data from 15 patients and show no systemic toxicity or significant immune reactions but some adverse events related to ocular surgery. There was a significant improvement in the full-field sensitivity testing in study eyes compared to control eyes. Three patients have also had vector administration to their second eye, 1.7–3.3 years after the initial treatment, with a subsequent follow-up reported after 6 months [19]. No detectable T-cell or neutralizing antibody response was found to the vector or transgene product. In terms of function, all “second” eyes showed improvement in full-field sensitivity testing, pupillometry, and functional MRI, suggesting that eventually treatment of both eyes may be safe and efficacious.
Clinical trial results have recently been published of phase 1/2 trial treating a further retinal degeneration called choroideremia, an X-linked recessive condition in which there are mutations in the CHM gene encoding Rab escort protein-1 (REP1). In this trial, an AAV2 vector expressing REP1 under the control of a CBA promoter was delivered subretinally in six patients. There were no adverse effects (related to both surgery and vector administration), and in two patients, visual acuity improvements were reported [20]. A further clinical trial to treat retinitis pigmentosa due to MERTK mutations is underway (NCT01482195), the results of which are awaited.
Gene therapy for retinal disease has so far proven to be safe and efficacious. There are numerous further retinal degenerations that may be treated using gene therapy for which new vectors will need to be developed and tested.
In this chapter, we will discuss features of AAV vector design to be considered when targeting retinal cells for gene therapy, protocols for vector production, and methods of testing vectors in vitro and in vivo.
2 Strategies for Vector Design
AAVs are single-stranded DNA parvoviruses that are not associated with pathogenicity in humans. Their 4.7 kb genome consists of two coding sequences, rep and cap, on either side of which lies a palindromic region known as an inverted terminal repeat (ITR) of 145 bp [21], required for replication, packaging, and insertion into the host genome [22]. The latter process occurs into a specific site on human chromosome 19 (AAV2) [23], but the majority of the wild-type AAV DNA exists as circular double-stranded episomes in the host cell [24]. This makes AAV more predictable than other viruses such as lentiviruses which may integrate randomly causing insertional mutagenesis. Recombinant AAV (rAAV) vectors for gene therapy are generated by deleting the entire coding sequence of the wild-type AAV genome and replacing it with a reporter gene such as the green fluorescent protein (GFP) or a therapeutic transgene. This eliminates the ability of the AAV vector to integrate into the host genome, further increasing its safety as a therapeutic vector.
2.1 Pseudotyping
Over 100 AAV serotypes have been described [25], and of these, AAV2, AAV3, and AAV5 are endemic to humans [26]. The molecular clone for AAV2 was the first to be isolated [27], and therefore, this virus and recombinant AAV2 have been most widely studied as a vector for gene therapy, showing efficient and long-term gene expression in a number of tissues. In order to further increase transduction efficiency and tropism to different cell types, vectors have been pseudotyped in which the ITRs of one serotype are packaged in the capsid of another serotype [28]. For example, by packaging AAV2 ITRs in an AAV5 capsid (generating a rAAV2/5 vector), this confers the safety and efficient expression of rAAV2 with the tropism of AAV5.
When deciding on a vector for rAAV gene therapy in the retina, the choice of pseudotype to use is important. The ability of pseudotyped vectors to transduce retinal cells has been extensively assessed in wild-type mice following both subretinal and intravitreal delivery to the eye; however, tropism in primates may vary from the murine models. rAAV2/2 and rAAV2/8 are both able to transduce ganglion cells following intravitreal injection [25], and therefore, one of these vectors should be considered if ganglion cells are the target for gene therapy. Following subretinal injection, rAAV2/1 and rAAV2/9 are able to transduce retinal pigment epithelium (RPE); rAAV2/2, rAAV2/5, rAAV2/7, and rAAV2/8 are able to transduce both RPE and photoreceptors [25]. Transduction kinetics and efficiency also vary between these vectors; for example, rAAV2/5 has a quicker onset with a gene expression evident at 5–7 days following subretinal injection in a wild-type mouse, when compared to rAAV2/2, in which gene expression is evident at 2 weeks. Studies have also demonstrated more efficient photoreceptor transduction with rAAV2/5 than rAAV2/2 [21].
2.2 Choice of Promoter
A further decision regarding vector design is the promoter to be used: whether to choose a ubiquitous promoter or restrict gene expression using a cell-specific promoter. Regarding ubiquitous promoters, the two most extensively assessed in retinal transduction are the CMV (human cytomegalovirus immediate-early gene) promoter and the CAG (comprising the CMV early enhancer and chicken β-actin (CBA) promoter), the latter being used in two of the human clinical trials for the treatment of Leber’s congenital amaurosis to drive RPE65 gene expression [15]. Following subretinal delivery, transgene expression in RPE cells, photoreceptor cells, and ganglion cells has been shown with both the CMV [29] and CAG promoters [13]. However, in other tissues, AAV-driven transgene expression has been demonstrated to be 137 times higher when using the CBA promoter compared to CMV [30]. In the brain [31] and muscle [32], there is evidence that the CMV promoter may be silenced due to methylation in the longer term, leading to transient or reduced gene expression.
For certain retinal degenerations, limiting gene expression to a specific cell type may have benefits with respect to safety and functional efficacy, although in some cases overall levels of transduction may be lower than with the use of a ubiquitous promoter. Regarding expression in the RPE, the 1.6 kb RPE65 promoter has been successfully used to limit gene expression to the RPE following subretinal delivery in Briard dogs [33] and has also been used in human clinical trials, although with questionable efficacy [14]. In targeting photoreceptors, the 292 bp human rhodopsin kinase (hGRK1) promoter has been shown to be effective in driving high levels of gene expression and specifically target rods and cones in both mouse models and nonhuman primates [34]. In conditions such as achromatopsia, it may be useful to limit gene expression to cone photoreceptors alone. For this purpose, the 2.1 kb PR2.1 red cone opsin promoter has been shown to specifically drive the gene expression in L/M cones in the normal canine retina [35], and the human cone arrestin promoter has shown expression in S and M cones in mice [36].
Optogenetic strategies for restoring vision in the degenerate retina might be optimized through gene delivery to bipolar cells. A 200 bp enhancer region from the murine GRM6 gene fused to a 203 bp SV40 (simian vacuolating virus 40) core promoter has been demonstrated to restrict transgene delivery to rod and cone bipolar cells following subretinal delivery to the rd10 mouse model of retinal degeneration [37]. In specifically targeting ganglion cells, the 2.8 kb human connexin 36 promoter produced a selective transduction of foveal ganglion cells following intravitreal delivery to the macaque retina [38], although the large size of this promoter may limit its use.
2.3 WPRE
The woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) facilitates nuclear export of viral RNA. Incorporation of this sequence into the 3′ untranslated part of the AAV genome has been demonstrated to increase gene expression by a factor of 1.8 times in brain tissue [31] and a sixfold increase in gene expression in cultured primary human fibroblasts compared to a vector which did not contain WPRE [39]. AAV vectors containing WPRE have been used in clinical trials in humans for the treatment of Parkinson’s disease (AAV-GAD study) [40].
Of note, the native WPRE contains a promoter and sequence for the first 60 amino acids of the woodchuck hepatitis virus X protein, which has been linked to the generation of liver tumors [41]. Therefore, modification of the WPRE sequence to prevent X antigen expression can be considered, and such a sequence has been used recently in the AAV vector administered in a clinical gene therapy trial to treat choroideremia (NCT01461213).
2.4 Self-Complementary Vectors
Following transfection by AAV, the onset of transgene expression is delayed by several days, and one factor contributing to this is the need to synthesize a complementary strand of DNA from the single-stranded AAV genome within the host cell nucleus. In order to bypass this step and improve AAV transduction, self-complementary AAV (scAAV) vectors have been developed. These vectors contain a single-stranded inverted repeat genome, with a wild-type ITR at one end and a mutated ITR in the center. After uncoating, the viral DNA folds to form a double-stranded molecule [42]. These vectors have been assessed in the retina and were found to have more rapid onset and more widespread gene expression following subretinal delivery in mice when packaged in rAAV2/2 [43], in rAAV2/8 [44], and in the canine retina in rAAV2/5 [45] when compared to their single-stranded AAV counterparts. However, the drawback of these vectors is their small packaging capacity, since both forward and reverse copies of the transgene of interest must be included within the 4.7 kb AAV vector.
2.5 Capsid Mutation
There is ongoing work to improve transduction efficiency by AAV and increase the number of cell types that can be targeted with these vectors. It is known that the AAV capsid is important for binding to cell surface receptors, which then leads to AAV internalization into the cell via endocytosis [46]. Recent work has shown that modification of the capsid VP3 protein, specifically point mutation of tyrosine residues to phenylalanine, can significantly increase transgene expression in HeLa cells in vitro and murine hepatocytes in vivo [47]. The exposed tyrosine residues on the capsid surface are believed to be involved in the ubiquitination pathway, which tags the protein for proteasome-mediated degradation. By modifying these residues, vector ubiquitination is thought to be decreased which leads to increased intracellular trafficking and more vector being delivered to the cell nucleus.
There are seven exposed tyrosine residues on the VP3 protein of the AAV capsid, and vectors containing mutations of one to all seven of these residues have been generated and assessed in the murine retina. Single mutations of capsid tyrosine residues in rAAV2/2, rAAV2/8, and rAAAV2/9 in self-complementary AAV vectors have been shown to significantly increase ganglion cell transduction when delivered intravitreally and RPE and outer retinal transgene expression when delivered subretinally in wild-type mice [48]. Transduction of cells in the inner nuclear layer such as bipolar cells, which are important for optogenetic strategies to restore vision, has previously proven to be difficult, possibly due to the viral particles being degraded before they have access to this layer. Tyrosine (Y) to phenylalanine (F) capsid mutations also appear to affect the ability of the AAV virus to penetrate the retina: a single mutation in rAAV2/8 (Y733F) has been demonstrated to result in bipolar cell transduction in the rd10 mouse model of retinal degeneration [37], and the rAAV2/2 quadruple and pentuple mutants are able to transduce bipolar cells when delivered either subretinally or intravitreally in wild-type mice [49]. Therefore, the ability to generate these capsid mutant vectors may allow more efficient transduction of the retina, targeting a wider range of cell types and requiring a lower titer of virus to be used which would have less risk of generating an immune response.
3 Gene Therapy of Large Genes Using AAV
The maximal packaging capacity of AAV is approximately 5 kb; however, many of the genes implicated in retinal degenerations exceed this size, such as ABCA4 mutations, which lead to some of the most common forms of inherited retinal degeneration. ATP-binding cassette, subfamily A, member 4 (ABCA4) is localized to the outer segments of rods and cones and acts as a membrane transporter for all-trans-retinal. If, due to a lack of functional ABCA4, all-trans-retinal is not transported out of the disk lumen efficiently, it reacts with phosphatidylethanolamine (PE) lipids in the disk membrane to form insoluble toxic products such as N-retinylidene-N-retinyl-ethanolamine (A2E), which get phagocytosed by the RPE cells. This disrupts RPE function and leads to cell death, which in turn results in a secondary photoreceptor degeneration. Being able to restore a functional copy of the ABCA4 gene to photoreceptors would have important therapeutic implications, but given the size of the ABCA4 (6.7 kb), gene therapy attempts require further considerations.
Lentiviral vectors have a larger packaging capacity than AAV and have shown some promise in ABCA4 gene therapy of the mouse knockout model [50]. However, the current abilities of lentiviral vectors to transduce photoreceptor cells are limited [51]. Studies investigating AAV vectors revealed that the optimal genome size for packaging is 4.1–4.9 kb, although larger genomes up to 5.2 kb were also detected [52]. Further evidence of AAV being able to package genomes as large as 6 kb was provided by Grieger and Samulski [53] who showed that transduction of vectors containing these larger genomes is enhanced by a treatment with proteasome inhibitors. This effect was also observed in mouse and dog models of hemophilia when treated with AAV vectors containing oversized genomes of 5.6 kb [54].
In 2008, Allocca et al. offered hope that AAV could package genomes of up to 8.9 kb [55]. Their data initially suggested that an Abca4 transgene was efficiently packaged within different AAV serotypes and effectively transduced Abca4 −/− mice. 8.9 kb transgene fragments were detected by a Southern blot analysis and full-length Abca4 isolated from injected Abca4 −/− retina. Despite these data, later studies revealed it was highly unlikely that AAV was packaging the full-length 8.9 kb transgene. The data observed were found to be due to a recombination of partially packaged transgenes within the host cells [56–58]. Strand-specific probes used to detect single-stranded AAV genomes revealed packaging that occurs primarily from the 3′ end until the capsid contains its maximal capacity of ssDNA [57]. Despite this finding, the above studies all detected the full-length expression cassette that exceeded the size actually packaged within the AAV. There are two proposed mechanisms for this: the first is that recombination occurs between overlapping homologous genome sequences and the second that sense and antisense genomes from two AAV vectors anneal with second strand synthesis occurring from their free 3′ ends, Fig. 2a. These potential mechanisms have been used to develop approaches for large gene therapy using AAV.
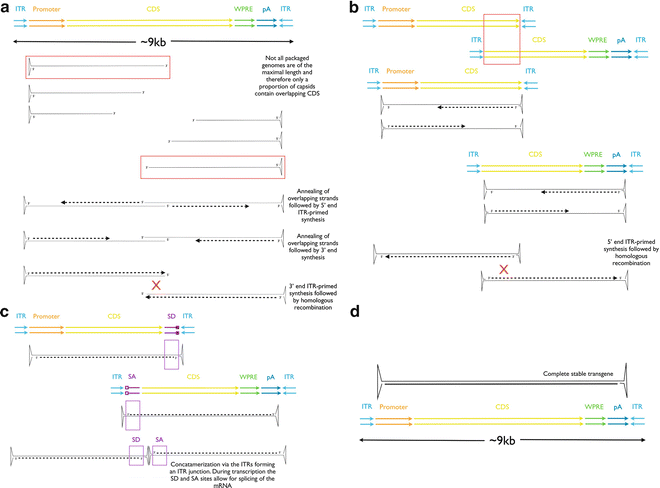
Fig. 2
Strategies for production of transgenes >5 kb. (a) The partially packaged strategy whereby the virus packages an undefined length of DNA from each 3′ end of the ITRs, resulting in capsids carrying varying lengths of genomes. Those containing overlapping segments have the potential to form the full-length transgene; (b) the overlapping strategy in which two defined fragments of the larger transgene are packaged between two ITRs, the resulting genomes undergo homologous recombination within the host cell to form the full-length transgene; (c) the trans-splicing approach in which the large transgene is again split into two defined fragments, but this time the generation of the full-length transgene depends on concatamerization between the two viral genomes following which the unwanted ITR junction is spliced out via the splice donor (SD) and splice acceptor (SA) sites
Partially packaged genomes containing only one ITR have been shown by several research groups to transduce host cells in vitro and in vivo with proof of principle demonstrated in the muscle [59] and lung [60] in addition to the retina [55]. An overlapping dual vector therapy of partially packaged large transgenes for dysferlinopathies has not only revealed in vivo production of the full-length 7.7 kb transcript and encoded protein, but this was also able to promote muscle membrane repair [61].
Overlapping approaches contain specifically generated fragments of the split coding sequence with a defined overlap region in each vector. The difference to the partially packaged approach is that two stable AAV genomes are generated with the split transgene contained within two ITRs. This approach has resulted in a sufficient cotransduction with subsequent homologous recombination of the overlapping transgenes to elicit a functional improvement in a mouse model of Duchenne muscular dystrophy [62]. That this dual vector approach has provided evidence of functional improvement in other tissue types and models of disease suggests the potential for such positive effects in the treatment of inherited retinal dystrophies are also plausible.
A variant on the overlapping approach is the trans-splicing method. This involves splitting the transgene into two ≤5 kb cassettes and packaging them separately, as in the alternative overlapping approach. The difference is the addition of splice donor/acceptor sites so that the first transgene carries a splice donor site following on from the split CDS and the second transgene carries a splice acceptor site prior to the split CDS. Following cotransduction, the AAV genomes are believed to undergo a recombination via the ITRs with the splice signals allowing for subsequent removal of the ITRs, creating a complete transgene exceeding 5 kb; proof of principle of the trans-splicing approach has been shown in muscle [63–66] and retina [67], though the relative effects compared to unsplit vectors are reduced, and efficiency may be as low as 7 % compared to the equivalent full-length single vector [59]. Of note, the overlapping approach also investigated in this study showed only 2.4 % efficiency compared to the respective full-length single vector. A further comparison of the overlapping and trans-splicing transgene approaches also revealed the overlapping approach to be the less effective with just one third of the activity compared to the single vector control, whereas the trans-splicing approach showed around half the activity of the single vector control [68].
A hybrid AAV dual vector approach has also been described containing highly recombinogenic sequences and splice donor/acceptor sites [68] and when tested showed no significant difference compared to the single AAV vector control. A more recent study has shown a hybrid trans-splicing/overlapping approach to be more effective in delivering large genes to photoreceptors than the overlapping approach. This method is an encouraging new development in the delivery of large genes using AAV as it offers two opportunities for the opposite transgenes to recombine, either by homologous recombination or by ITR concatamerization [69].
Cotransduction efficiency is particularly important when using dual AAV vectors [70]. In the overlapping approach, transgene overlap sequence and AAV serotype play important roles in effective treatment [71]. An ~1 kb overlap has been shown to be effective in inducing a homologous recombination [59]. However, the use of a highly recombinogenic sequence of 0.44 kb allowed for smaller bridging sequences to be included in the vector in place of a large overlap. For the trans-splicing approach, the larger bridging sequence of 0.87 kb was twice as effective in generating expression of the recombined transgene [72]. Correct serotype selection has also been shown to enhance the dual vector strategy in different tissue types including the lung [60], muscle [71], and retina [67].
Despite the packaging limitation of AAV vectors, the likelihood of being able to provide effective gene therapy for inherited diseases caused by mutations in large genes is now looking promising with these dual vector approaches.
4 Virus Production: Methodology
Production of AAV involves co-transfecting a stable cell line such as HEK 293T with three plasmids: (1) containing AAV ITRs and the reporter or therapeutic gene of interest, (2) containing the AAV rep and cap genes, and (3) containing helper genes identified from adenovirus (helper plasmid). Alternatively, plasmids can now be purchased from companies such as the PlasmidFactory that carry both the helper and AAV rep/cap genes, allowing for a dual-transfection strategy using this and the plasmid (1) above.
4.1 Cloning and Transformation
A typical example of a plasmid containing a GFP transgene between AAV ITRs is shown in Fig. 3. The recombinant expression cassette of choice should be inserted into the plasmid using appropriate restriction sites and ligated using DNA ligase. If the same restriction sites are required at each end of the transgene insert, then the digested plasmid backbone should be treated with alkaline phosphatase prior to ligation with the insert.
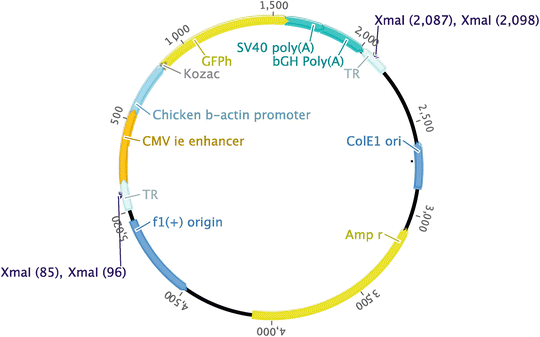
Fig. 3
Cloning map. Example of a plasmid containing AAV inverted terminal repeats with a green fluorescent protein coding sequence. Xma I restriction sites are annotated
In its native form, the 145 bp AAV ITR forms a stable secondary structure (Fig. 4) and is therefore susceptible to deletion in the cloning process. For this reason, we recommend the use of XL10-Gold Ultracompetent bacteria (Agilent Technologies) for cloning procedures, which we have found to be reliable at preserving ITR integrity.
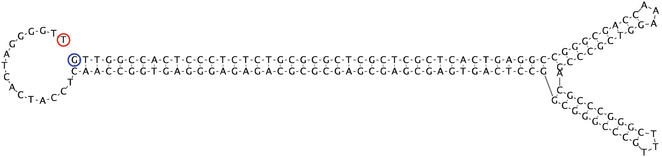
Fig. 4
Inverted terminal repeat (ITR). Secondary structure formed by palindromic sequence of ITR. Image generated using Geneious software: Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A (2012) Geneious v5.6, available from http://www.geneious.com
4.1.1 Transformation Protocol
This is based on the manufacturer’s guidelines. Thaw 75 μl XL10-Gold bacteria on ice and add 3 μl β-mercaptoethanol, and gently mix every 2 min for 10 min. Incubate the cells with 5 μl of the ligation mixture on ice for 30 min. Heat shock at 42 °C for 30 s and then incubate for 2 min on ice. Add 925 μl of SOC Medium (Invitrogen) and incubate at 37 °C in a shaker rotating at 225 rpm for 1 h. Spread 200 μl of the transformation mix on LB agar plates containing 100 μg/ml ampicillin, and incubate at 37 °C for 16 h. Pick single colonies, culture in 4-ml LB/ampicillin for 16 h, and isolate the plasmid using a Miniprep Kit (Sigma-Aldrich). To generate the high concentrations of plasmid required for virus preparation, culture an isolated plasmid (fully sequenced for confirmation of the transgene insertion) in 2 l of LB/ampicillin and isolate using a Megaprep Kit (QIAGEN).
4.2 Checking ITRs
Due to the nature of the secondary structure formed by AAV ITRs, we have found that confirmation of their presence following cloning is difficult. Direct sequencing and isolation via polymerase chain reaction (PCR) both give variable and often incomplete results. Therefore, we have found that the most accurate method of confirming the presence of ITRs is via a restriction digest.
4.2.1 Method
Two Xma I restriction sites are present within the ITR sequence (Fig. 3); check that this enzyme does not cut anywhere else in the expression cassette inserted. If the distance between the two ITRs is approximately half the plasmid size, a second restriction enzyme with only one recognition site between the ITRs will also be required.
Incubate 500 ng plasmid with 1 μl Xma I, 2.5 μl buffer, and 2.5 μl BSA and make up to 25 μl with sterile water for 2 h at 37 °C. Separate the products using agarose gel electrophoresis, and the size of bands seen will indicate whether the clones are positive for both ITRs and the inserted transgene.
4.3 Transfecting Cells for Virus Production
Transfecting the large number of cells needed for virus production is most easily done in a HYPERFlask (Corning). This is a multilayered flask with a total growth area of 1,720 cm2.
Seed a 75 cm2 tissue culture flask with 17 × 106 HEK 293T cells in Dulbecco’s Modified Eagle Medium (DMEM) with 4.5 g/l glucose, 2-mM l-glutamine, 10 % fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Each flask will then seed one HYPERFlask. We recommend using two HYPERFlasks and therefore seeding two flasks at the above concentration per virus to be made.
After approximately 72 h, or when the cells are 80 % confluent, loosen the cells and resuspend in 200 ml media. Gently transfer the cells from one 75 cm2 flask into a HYPERFlask. This is most easily done with the HYPERFlask tilted to about 60°, so the cells are divided between all layers of the HYPERFlask. Then, turn the HYPERFlask to an upright position and fill completely with media (an additional 300–350 ml) until the flask is full to the lowest thread on the neck. Gently tap the flask to dislodge any trapped air bubbles.
After 72 h, transfect the cells in each HYPERFlask with a total of 500 μg DNA. The ratio of helper plasmid to rep/cap plasmid to transgene plasmid is dependent on the size of the plasmids. Add the size of each plasmid to provide a total in kb. Divide 500 (the amount of total DNA required) by that total to give the ratio value (R). Multiply each plasmid size by the R to give the amount in μg of each plasmid to be used (Table 1). Then, determine the volume in μl required of each plasmid that provides the respective amount of DNA.
Table 1
Calculating ratio of plasmids for virus production
Plasmids | Size (kb) | Amount per transfection (μg) |
|---|---|---|
pHelper | 18.7 | 18.7 × R |
pRep-cap | 7 | 7 × R |
pTransgene | t | t × R |
Total | K kb | 500 μg |
Ratio (R) mass/kb | 500/K |
Measure the three plasmids into a 50-ml Falcon tube, and make up to 10 ml with 150-mM NaCl. To make a 150-mM sodium chloride solution, dilute 4.38 g NaCl in 500 ml dH2O. In a separate Falcon tube, measure out 1.125 ml 10 mM polyethylenimine (PEI) transfection reagent (linear polyethylenimine, MW 25,000, Polysciences, 23966-2), and make up to 10 ml with 150 mM NaCl.
Add the PEI drop by drop into the Falcon tube containing the DNA, gently swirling the mixture continuously while doing so. If the PEI is added too quickly, precipitation will occur and the contents of the tube will go cloudy. Incubate the mixture at room temperature for 20 min. Add this to a 500 ml bottle of DMEM containing 4.5 g/l glucose, 2 % fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Slowly drain the old media from the HYPERFlask and fill with media containing the DNA and transfection agent as prepared above. Top up the HYPERFlask with additional media if required, ensuring again there are no air bubbles. Incubate at 37 °C for 72 h.
Related posts:
 Gene Therapy Approaches to Promoting Axonal Regeneration After Spinal Cord Injury
Gene Therapy Approaches to Promoting Axonal Regeneration After Spinal Cord Injury
 Gene Therapy Approaches Using Reproducible and Fully Penetrant Lentivirus-Mediated Endogenous Glioma Models
Gene Therapy Approaches Using Reproducible and Fully Penetrant Lentivirus-Mediated Endogenous Glioma Models
 Gene Therapy for Huntington’s Disease
Gene Therapy for Huntington’s Disease
 Gene Therapy for Epilepsies
Gene Therapy for Epilepsies
 Gene Therapy for Chronic Pain: How to Manipulate and Unravel Pain Control Circuits from the Brain?
Gene Therapy for Chronic Pain: How to Manipulate and Unravel Pain Control Circuits from the Brain?
 Adeno-Associated Vectors for Gene Delivery to the Nervous System
Adeno-Associated Vectors for Gene Delivery to the Nervous System
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


