Chemical class
Toxic molecule
Animal models
Supporting evidence from human ALS cases
Reference
Amino acids
Glutamatea
hSOD1G85R mice hSOD1G93A mice
TDP-43M337V rats
TBPH depleted or over-expressing Drosophila
Defective uptake in synaptosomes prepared from ALS patients due to selective down-regulation of EAAT2
Increased levels in the CSF of ALS patients.
D-serine
hSODG93A mice
Mutation in D-amino acid oxidase in a fALS pedigree.
Pro-inflammatory mediators
PGD2
hSODG93A mice: co-cultures of astrocytes and motor neurons from embryonic stem cells
(Di Giorgio et al. 2008)
Interferon γ
hSODG93A mice
(Aebischer et al. 2011)
Growth factors
TGFβ
hSODG93A mice
(Phatnani et al. 2013)
Pro-NGF
hSODG93A mice
Increased levels in the CSF of ALS patients.
(Ferraiuolo et al. 2011b)
Other
Lipocalin 2
Mutant SOD1, mutant TDP-43, mutant FUS rats
As yet, the mechanisms leading to EAAT2 depletion in ALS have not been fully elucidated, although several mechanistic hypotheses have been postulated. Initially, it was reported that the loss of this transporter was the consequence of aberrant splicing of its messenger RNA (mRNA) (Lin et al. 1998) . Yet, this event was later proved unspecific for ALS cases, as it was observed also in control and Alzheimer’s Disease patients (Honig et al. 2000) . EAAT2 was subsequently proposed to be a substrate of caspase-3 (Boston-Howes et al. 2006) , an enzyme executing the proteolytic phase of apoptotic programmed cell death. Cleavage generates a C-terminal fragment, and leads to inhibition of the transporter activity (Boston-Howes et al. 2006). The product of EAAT2 cleavage was also described to undergo additional post-translational modification, namely SUMOylation (Gibb et al. 2007) , and to accumulate in spinal astrocytes from hSOD1G93A mice at the symptomatic stage (Foran et al. 2011) . More recently, an additional theory has been proposed, based on the occurrence of aberrant editing events in intron 7 of the EAAT2 pre-mRNA in the affected areas of ALS patients (Flomen and Makoff 2011) . Such events appears to lead to alternative polyadenylation, ultimately causing premature transcriptional termination and reduced levels of EAAT2 (Flomen and Makoff 2011). Down-regulation of EAAT2 was also suggested to be the result of defective signalling from neurons. For example, presynaptic terminals were shown to transcriptionally control the expression of EAAT2 by modulating the astrocytic levels of the nuclear factor Kappa-B Motif-Binding Phosphoprotein (KBBP). The loss of presynaptic signalling, following axonal degeneration, resulted in diminished EAAT2 expression (Yang et al. 2009) . Finally, experiments in culture suggested that neuronal cells have the capacity to release exosomes containing the microRNA 124a (miR-124a) (Morel et al. 2013) . Such organelles can be internalized by astrocytes, where they increase the astrocytic miR-124a. In turn, the latter up-regulates EAAT2 expression through a yet unidentified mechanism (Morel et al. 2013). Since miR-124a expression is down-regulated in the spinal cord of hSOD1G93A mice, it was speculated that its reduced expression may be responsible for the diminished levels of EAAT2 in vivo (Morel et al. 2013). While this amount of evidence globally failed to provide definitive conclusions about the mechanism driving EAAT2 loss in ALS, it prompted the idea that the relevance of the transporter in ALS may be determined by directly modulating its expression in vivo. This hypothesis was pursued by genetic and pharmacological approaches. Thus, transgene-driven expression of EAAT2 in the hSOD1G93A mouse line was shown to delay neurodegeneration and disease onset (Guo et al. 2003) . Furthermore, double transgenic mice obtained by crossing hSOD1G93A ALS mice with animals showing ubiquitous over-expression of the Peroxisome Proliferator-Activated Receptor γ Coactivator 1α (PGC1α) exhibited enhanced EAAT2 expression, and this correlated with improved motor function (Liang et al. 2011) . From a pharmacological standpoint, the screening of a panel of US FDA-approved drugs revealed that beta-lactam antibiotics, and ceftriaxone in particular, are capable to enhance EAAT2 expression (Rothstein et al. 2005) . Chronic administration of ceftriaxone in hSOD1G93A transgenic mice extended the animal lifespan (Rothstein et al. 2005), thus raising great hope that this compound may successfully control disease progression in ALS patients. It is to mention that a clinical trial with ceftriaxone is currently ongoing, and results from Phase I and II indicate that this drug is generally well tolerated and can successfully reach the CNS at therapeutic doses (Berry et al. 2013) . In an attempt to identify other compounds that enhance EAAT2 expression, a further screening was undertaken by the same group, which led to the identification of harmine (Li et al. 2011) , a beta-carboline alkaloid, as a promising agent to be tested in vivo, in preclinical trials.
Further support to the excitotoxic hypothesis of ALS was provided by the landmark observation that mutant SOD1-expressing astrocytes can release high levels of D-serine, a co-activator of the N-methyl-D-aspartate (NMDA) receptors, and this exacerbates glutamate toxicity on motor neurons in vivo (Sasabe et al. 2007, 2012) . The relevance of this amino acid was corroborated also by the identification, in a fALS pedigree, of a unique mutation (Arg199→Trp substitution) in the gene coding for D-amino acid oxidase (DAOR199W), an enzyme that regulates the levels of D-serine (Mitchell et al. 2010) . In vitro characterization revealed that this mutation not only causes the loss of DAO enzymatic activity, but also promotes both the formation of ubiquitin aggregates and apoptosis in neuronal cells. Remarkably, the detrimental effect of the mutant protein was observed even when motor neurons were co-cultured on DAOR199W-expressing astrocytes, thus suggesting that the neurodegenerative process can be mediated by both cell autonomous and non-cell autonomous events (Mitchell et al. 2010).
Increased production of reactive oxygen species (ROS), following mitochondrial dysfunction of mutant hSOD1G93A astrocytes, was also counted among the mechanisms contributing to motor neuron sufferance (Fig. 11.1). Oxidant species and Nitric Oxide Synthase (NOS) products were proposed to be involved in the neurotoxic process in vitro (Cassina et al. 2008) . In keeping, the over-production of ROS by human astrocytes expressing the hSOD1G37R mutant protein was corroborated by the observation that activation of NADPH oxidase (NOX2) contributes to the establishment of a condition of oxidative stress (Marchetto et al. 2008) . Interestingly, pharmacological inhibition of NOX2 activity by the drug apocynin reversed both ROS production and motor neuron cell death driven by mutant SOD1-expressing astrocytes (Marchetto et al. 2008). In addition, pharmacological (Cassina et al. 2008) or genetic (Vargas et al. 2008) manipulations apt to support the mitochondrial activity of astrocytes (Miquel et al. 2012) , or to potentiate the antioxidant defenses of the cells, (Vargas et al. 2008) showed a neuroprotective potential, thus establishing a direct link between the astrocytic production of free radicals and the process of neuronal cell death.
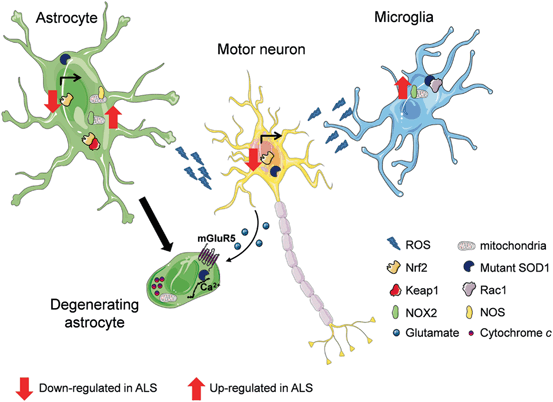
Fig. 11.1
Impact of glial cells on motor neuron sufferance in ALS-SOD. In normal conditions, healthy astrocytes support the welfare of motor neurons by supplying trophic and metabolic factors as well as by scavenging potentially harmful agents. The transcription factor Nrf2 mediates the expression of several antioxidant proteins. Normally, its activity is repressed by the Keap1 protein in cytoplasm. However, when cells are exposed to chemical or oxidative stress, Nrf2 escapes Keap1-mediated repression, translocates into the nucleus, and promotes the expression of enzymes involved in the detoxification response. In the presence of mutant SOD1, the Nrf2-Keap1 pathway is down-regulated, and both oxidant species and Nitric Oxide Synthase (NOS) products are release from astrocytes following mitochondrial dysfunction (top, in green). During ALS, mutant SOD1 expression greatly impacts on the astroglial function, and some astrocytes (bottom, in green) display an enhanced susceptibility to physiological concentrations of the transmitter glutamate. This latter triggers a gliodegenerative process via the activation of its metabotropic receptor type 5 (mGluR5). mGluR5 activation induces aberrant Ca2+ release from the intracellular stores, mitochondrial disarrangement with cytochrome c release, and apoptotic cell death. In microglial cells (in blue), the activation of NADPH oxidase (NOX2) participates to the establishment of a condition of oxidative stress. SOD1 normally contributes to the regulation of NOX2 activity by binding to its regulatory protein Rac1. In the presence of mutant SOD1, a stronger interaction with Rac1 is established, and this leads to aberrant NOX2 activation with the consequent over-production of reactive oxygen species
The idea that targeting oxidative stress in astrocytes might be an efficient therapeutic option for ALS was further corroborated by a number of recent studies focused on the nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2 is a transcription factor that mediates important cellular defense mechanisms against oxidative stress, through its capacity to regulate the expression of an array of antioxidant proteins. Under normal condition, Nrf2-dependent transcription is repressed by the inhibitor Kelch-like ECH-associated protein 1 (Keap1), which sequesters Nrf2 in the cytoplasm and promotes its degradation by the ubiquitin proteasome pathway. However, when cells are exposed to chemical or oxidative stress, Nrf2 escapes Keap1-mediated repression and translocates into the nucleus. This allows its interaction with the antioxidant response element (ARE), a cis-acting regulatory sequence located in the promoter region of a number of genes. By this means, Nrf2 promotes the expression of enzymes involved in the detoxification and antioxidant response (Bryan et al. 2013) . The fact that the Nrf2-Keap1 signalling pathway may be involved in neuroprotective mechanisms in ALS was initially suggested by multiple investigations in vitro , in motor neuronal cell culture systems expressing either hSOD1G93A or various TDP-43 mutants (Kirby et al. 2005; Pehar et al. 2007; Duan et al. 2010) . Similar conclusions arose from additional studies investigating the expression of Nrf2 and Keap1 in post-mortem tissue samples from ALS patients (Sarlette et al. 2008) and hSOD1G93A transgenic animals (Mimoto et al. 2012) . Thus, the impact of different compounds acting on this signalling cascade was investigated in the hSOD1G93A animal model of the disease. Among the activators of the Nrf2/ARE system taken into consideration, there are DL-3-n-butylphthlide (Feng et al. 2012) , S[+]-Apomorphine (Mead et al. 2013) , and triterpenoids (Neymotin et al. 2011) . While all of these molecules resulted effective in improving the animal motor performance (Neymotin et al. 2011; Feng et al. 2012; Mead et al. 2013), the overall impact on survival was either modest (Neymotin et al. 2011; Feng et al. 2012) or even null (Mead et al. 2013). Consistent with this, genetic approaches apt to modulate neuronal Nrf2 expression in vivo, in the hSOD1G93A and hSOD1G85R mouse models, moderately delayed disease onset without affecting the animal lifespan (Vargas et al. 2013) . Similarly, a gene therapy protocol apt to induce Nrf2 over-expression in neurons failed to mitigate the detrimental phenotype in hSOD1G93A ALS mice (Nanou et al. 2013) . At variance with these results, Nrf2 activation in astrocytes gave more promising results. Firstly, increasing Nrf2 activity in astrocytes, by transfection or pharmacological methods, resulted neuroprotective in astrocyte-motor neuron co-culture experiments (Vargas et al. 2005, 2006). Secondly, transgene-driven expression of Nrf2 in astrocytes prevented motor neuron degeneration, delayed disease onset, and extended survival of hSOD1G93A ALS mice (Vargas et al. 2008) (Fig. 11.1).
Although not all studies agree (Guo et al. 2013) , these reports suggest that the Nrf2-Keap1 pathway may represent an attractive pharmacological target for therapeutic intervention in ALS. More importantly, they emphasize the concept that Nrf2 activation should be selectively achieved in the astrocytes.
Besides glutamate and oxidant species, additional neurotoxic candidate molecules of astrocytic origin were indicated in different inflammatory mediators . For example, abnormal signalling in astrocytes from different prostaglandins (PGs) was initially proposed as a likely contributor to motor neuron demise (Di Giorgio et al. 2008) . Analysis of the expression of the PGD2 and PGE2 receptors in fact revealed that both proteins are up-regulated in hSOD1G93A astrocytes (Di Giorgio et al. 2008; Liang et al. 2008) . Besides, the relevance of the PGE2 EP2 receptor was directly investigated in vivo. Importantly, its genetic ablation was reported to limit the pro-inflammatory response, to delay the onset of motor impairment, and to extend the lifespan of hSOD1G93A ALS mice (Liang et al. 2008). An abnormal signal transduction pathway, leading to motor neuron cell death, was identified also upon the release of interferon γ(IFNγ) from hSOD1G93A astrocytes in co-culture experiments (Aebischer et al. 2011) . More recently, an investigation of the gene expression profile of mutant SOD1-expressing astrocytes identified striking changes even in a panel of growth factors. Thus, analysis of hSOD1G93A astrocytes and motor neurons in co-culture revealed a complex and interconnected alteration in the transcriptome of these cells, suggesting a potential role for Transforming Growth Factor β (TGFβ) signalling in ALS (Phatnani et al. 2013) . Furthermore, an up-regulation of the TGFβ type II receptor (TGFβ–RII) was described in motor neurons from hSODG93A mice, with a peak of expression occurring before the onset of the disease. Interestingly, such increase in TGFβ-RII immunoreactivity temporally paralleled the progression of reactive astrocytosis, as indicated by the presence of thicker and hypertrophic astrocytes around TGFβ-RII-immunopositive motor neurons (Phatnani et al. 2013) . Transcription profile analysis of astrocytes isolated by laser-capture microdissection from the lumbar spinal cord of pre-symptomatic hSOD1G93A mice further revealed an up-regulation of Nerve Growth Factor β (β-NGF) (Ferraiuolo et al. 2011b) , a molecule that triggers apoptotic cell death in the neighbouring motor neurons through the activation of the p75 neurotrophin receptor (Pehar et al. 2007) . An increased release of β-NGF from astrocytes expressing the mutant hSOD1G93A protein was confirmed in vitro, and immunodepletion of this peptide was shown to rescue the neurotoxic effect of hSOD1G93A astrocytes in co-cultures (Ferraiuolo et al. 2011b). It should be, however, mentioned that the contribution of glial cells to motor neuron degeneration in vitro was not only shown in those forms of the disease linked to SOD1 mutations. In fact, TDP-43 was reported to interact with the Nuclear Factor-kB (NF-kB), a master regulator of several genes involved in the inflammatory response, in both neuronal and glial cells (Swarup et al. 2011) . The direct consequence of this event is that, under stress conditions, TDP-43-expressing microglia and astrocytes produce enhanced pro-inflammatory cytokines as well as neurotoxic mediators (Swarup et al. 2011). A role for the astroglial NF-kB signalling complex in mediating the inflammatory response was further supported by a recent gene expression study made on astrocytes from familial and sporadic ALS patients (Haidet-Phillips et al. 2011) .
Among the reported neurotoxic factors secreted by ALS astrocytes, there is also lipocalin 2 (lcn2), a protein characterized by the ability to bind and transport lipids and other hydrophobic molecules. While lcn2 is involved in diverse cellular processes, there is considerable evidence suggesting that this protein is able to induce cell death by stimulating an apoptotic program (Devireddy et al. 2001, 2005) . Remarkably, Bi and colleagues recently reported that lcn2 is released from reactive astrocytes in cultured organotypic brain slices from transgenic rats with neuron-specific expression of mutant human TDP43M337V. Furthermore, in the brain of transgenic rats expressing mutant forms of TDP-43, FUS, or SOD1, this protein was strongly induced in reactive astrocytes (Bi et al. 2013; Tong et al. 2013) . In vitro, lcn2 is cytotoxic to primary neurons, and toxicity is enhanced in cells that express disease genes, such as mutant FUS or TDP-43 (Bi et al. 2013).
Altogether, these observations suggest that the dynamic interactions between astrocytes and motor neurons become impaired at different levels during ALS progression.
11.3.2 Astrocyte Dysfunction and Sufferance
Several lines of evidence indicate that some of the neurosupportive functions of the astrocytes can be lost during the progression of the disease . As mentioned in the previous paragraph, astrocytes were consistently reported to exhibit a dysfunction of the glutamate uptake system in ALS patients and transgenic rodents, and this was proposed to contribute to excitotoxic motor neuron cell death (Rothstein et al. 1992, 1995; Bruijn et al. 1997; Howland et al. 2002; Guo et al. 2003; Pardo et al. 2006) . In addition, astroglia were reported to modulate the intrinsic susceptibility of motor neurons to glutamate by fine-tuning the expression of the GluA2 subunit of the glutamatergic α-amino-3-hydroxy-5-methyl-isoxazole propionate receptors (AMPARs) (Van Damme et al. 2007) . Remarkably, the presence of GluA2 in the AMPAR subunit composition critically impacts the biophysical properties of the receptor and determines its permeability to calcium ions (Ca2+). In particular, the expression of GluA2 normally renders AMPARs impermeable to Ca2+, because of an RNA editing event of the Q/R site (Q, unedited; R, edited) in the GluR2 mRNA (Sommer et al. 1991) . Yet, astrocyte expressing the mutant hSOD1G93A protein appear to lose their capacity to modulate the expression of the GluA2 subunit of the AMPARs in the neighbouring motor neurons, thereby exposing them to enhanced Ca2+ influx and cell death (Van Damme et al. 2007). Besides their involvement in excitotoxic mechanisms, ALS astroglia were described to hold additional functional deficits, including mitochondrial defects as well as a reduced capacity to release the metabolic substrate lactate (Cassina et al. 2008; Ferraiuolo et al. 2011b) . Furthermore, the early observations of ubiquitinated SOD1- and active caspase-3-immunopositive inclusions in these cells, in the two hSOD1G93A and hSOD1G85R mouse models of the disease, raised the original idea that mutant SOD1 can be toxic to astrocytes themselves (Bruijn et al. 1997; Pasinelli et al. 2000) . In subsequent experiments, our group investigated the significance of such astrocytic inclusions in hSOD1G93A– expressing transgenic mice. We found that these unusual cells are specifically located in the motor neuronal microenvironment in the ventral horns of the spinal cord (Fig. 11.2) . Furthermore, they display morphological and biochemical features reminiscent of degenerating cells, including an atypical spheroid morphology with an increased diameter and a reduced number or even the absence of cell processes immunopositive for the Glial Fibrillary Acid Protein (GFAP) (Rossi et al. 2008; Martorana et al. 2012) . Dying astrocytes were first observed at the pre-symptomatic stage (Rossi et al. 2008), i.e. at a time when motor neurons exhibit axonal damage, but their somas are still vital (Pun et al. 2006) . The number of spheroid astrocytes significantly increased concomitant with the onset of neuronal degeneration and the appearance of ALS symptoms (Rossi et al. 2008; Martorana et al. 2012). The relevance of this phenomenon was more recently reinforced in the context of the human disease by the identification of cells with an analogous aberrant morphology in the spinal cord of ALS patients (Martorana et al. 2012). Interestingly, we realized that degenerating astrocytes, in the spinal cord of hSOD1G93A mice, were surrounded by glutamatergic terminals (Fig. 11.2), as to indicate that these cells can sense the neurotransmitter glutamate spilled over during synaptic activity (Rossi et al. 2008; Martorana et al. 2012) . Mechanistic studies in vitro revealed that astrocytes expressing different SOD1 mutations, either G93A or G85R, displayed an enhanced susceptibility to physiological concentrations of glutamate. The glutamatergic mechanism responsible for the deleterious event was found to involve metabotropic glutamate receptor 5 (mGluR5) signalling, which triggers an apoptotic gliodegenerative pathway in culture (Rossi et al. 2008) . More recently, the signalling prompted by the activation of group I mGluRs, including mGluR5, was further characterized .

Fig. 11.2
Degenerating astrocytes appear in the hSOD1G93A ALS mouse model at the pre-symptomatic stage of the disease. Arrows show ubiquitin-positive (in blue) astrocytes labelled with the astroglial marker Glial Fibrillary Acidic Protein (GFAP, in green). Degenerating astroglial cells are located in the motor neuronal microenvironment and became visible early during disease progression in the lumbar spinal cord of hSOD1G93A mice. They are enclosed by glutamate-containing pre-synaptic terminals immunolabelled for the Vesicular Glutamate Transporter 1 (VGLUT1, in red). Arrowheads indicate VGLUT1-positive glutamatergic vesicles and asterisks show motor neuronal somas. Scale bar, 20 μm
In normal astroglial cells, the activation of group I mGluRs is well known to cause the formation of inositol 1,4,5 trisphosphate (IP3), followed by IP3-mediatedrelease of Ca2+ from the endoplasmic reticulum (ER) stores, which results in intracellular Ca2+ oscillations (Zur Nieden and Deitmer 2006; Gunnarson et al. 2009) . Yet, mutant hSOD1G93A-expressing astrocytes responded to the receptor stimulation with an aberrant and persistent Ca2+ release from the intracellular stores. This correlated with mitochondrial disarrangement, cytochrome c release from mitochondria , and astrocyte degeneration (Martorana et al. 2012). These results are in line with the mitochondrial dysfunction previously described by Cassina and colleagues in mutant SOD1-expressing astrocytes (Cassina et al. 2008) .
The anti-apoptotic members of the Bcl-2 family, particularly Bcl-2 and Bcl-XL, were largely reported to confer cell death resistance by fine-tuning intracellular Ca2+ signalling through direct interaction with the IP3 receptor (IP3R) channels (Chen et al. 2004; White et al. 2005; Zhong et al. 2006; Li et al. 2007; Rong et al. 2008, 2009) . Interestingly, structure-function analysis of Bcl-2 homologues revealed that their N-terminal homology domain 4 (BH4) is essential for inhibition of apoptosis (Hunter et al. 1996; Lee et al. 1996; Huang et al. 1998) . Our group thus investigated the impact of the BH4 domain of Bcl-XL on astrocyte Ca2+ signalling by exploiting a biologically active BH4 peptide fused to the protein transduction domain of the HIV-1 TAT protein (TAT-BH4). We realized that TAT-BH4 modulates the IP3R-dependent Ca2+ release from the ER, and restores spontaneous Ca2+ oscillations in mutant SOD1-expressing astrocytes. This tight control of IP3Rs by the peptide prevents group I mGluR-driven aberrant release of Ca2+ from the intracellular stores, precludes the release of cytochrome c from mitochondria, and protects the cells from excitotoxic damage (Martorana et al. 2012) . Furthermore, chronic treatment of hSOD1G93A transgenic mice with TAT-BH4 reduces degeneration of spinal cord astrocytes and shows a positive impact on the disease manifestations (Martorana et al. 2012) .
Consistent with a specific vulnerability of ALS astrocytes, functional astroglia deriving from human induced pluripotent stem cells (iPSC) expressing the mutant TDP-43M337V protein were recently generated and shown to exhibit reduced cell survival (Serio et al. 2013) . Yet, in co-culture experiments, such cells were not harmful to wild-type motor neurons, possibly because in vitro differentiation may not have captured some of the detrimental properties that diseased astroglia harbor in vivo (Serio et al. 2013). In line with this conclusion, in transgenic rats expressing the same mutant form of TDP-43 in astrocytes, the loss of unhealthy astroglial cells paralleled progressive degeneration of motor neurons, denervation atrophy of skeletal muscles, and progressive paralysis (Tong et al. 2013) . Taken together, these findings support the view that glial cell sufferance can play a role in motor neuron degeneration also in ALS-TDP, though the evidence in this clinical subtype of the disease is still limited .
Another important function of the astrocytes that is worth mentioning concerns their association with the BBB, a complex structure tightly regulating the exchange of molecules between the brain parenchyma and the blood stream (reviewed in Daneman 2012) . On a structural standpoint, the BBB is made of endothelial cells, astroglia , pericytes and neurons, which globally establish a “neurovascular unit”. In this context, astrocytes project off their processes, and their endfeet ensheath the blood vessels. This strategic location suggests that they can importantly contribute to the maturation and maintenance of the BBB. Consistent with this vision, multiple studies in vitro have provided consistent evidence that astrocytes can influence the transendothelial resistance, a measure of BBB permeability; the organization of tight junctions between brain endothelial cells; and the luminal polarization of transporters, such the glucose transporter Glut-1 and the P-glycoprotein (Dehouck et al. 1990; Rubin et al. 1991; Hayashi et al. 1997; Sobue et al. 1999; Al Ahmad et al. 2011) . Although the integrity of the BBB is of outstanding importance for the maintenance of the optimal neuronal microenvironment, alterations of this structure have been described in a number of neurological disorders, including ALS (Daneman 2012). Early studies in the hSOD1G93A ALS mice identified ultrastructural damage to astrocytes and endothelial cells, leading to vascular leakage of intravenously injected Evans Blue (EB) dye already at the pre-symptomatic stage (Garbuzova-Davis et al. 2007) . EB extravasation abnormalities were found in both the cervical and lumbar spinal cord (Garbuzova-Davis et al. 2007), and correlated with altered expression of critical proteins for the regulation of the efflux of catabolites from the brain parenchyma, such as Glut-1 and P-glycoprotein (Garbuzova-Davis et al. 2007). Evidence of BBB damage and microhemorrhages was later extended to other transgenic mice, harboring the G37R and G85R SOD1 variants (Zhong et al. 2008) , and to sporadic ALS cases (Garbuzova-Davis et al. 2012; Winkler et al. 2013) . Given the importance of the BBB in tuning the access of drugs to the CNS, these findings should be taken in consideration when designing new therapies for ALS.
In conclusion, taken together, these studies confirm a multifaceted role for astrocytes in ALS, even though they fail to provide conclusive evidence as to whether the multiple and diverse contributions of astroglia are due to distinct astrocyte subpopulations or to the same population receiving different external inputs (Molofsky et al. 2012; Oberheim et al. 2012) . We suggest that a better understanding of the impact of the distinct astrocytic mechanisms to ALS pathogenesis will be certainly beneficial to develop targeted therapeutics .
11.4 Microglia
Microglia are the glial cell population deputed to the immune surveillance of the CNS. They typically respond to injury or disease with a massive activation in areas affected by neuronal degeneration. This condition is commonly known as “reactive microgliosis”. Reactive microglia have the capacity to release a wide variety of substances that can eitherlimit or exacerbate neuronal sufferance (reviewed in Aguzzi et al. 2013) . In both autoptic ALS cases and animal models, microgliosis was early recognized as a typical hallmark of the disease (Hall et al. 1998) . Yet, it was only with the recent advent of modern imaging technologies, coupled to the development of radiolabelled ligands, that the extent of this phenomenon could be assessed in vivo, in ALS patients, at the time of disease diagnosis. A number of positron emission tomography (PET) studies has been thus performed by neuroimaging the 18 kDa translocator protein (also known as the “peripheral benzodiazepine receptor”) (Turner et al. 2004; Corcia et al. 2012) , which is highly expressed in phagocytic inflammatory cells, including activated microglia (Papadopoulos et al. 2006) .
Several investigations, using mutant SOD1-expressing cellular and animal models of the disease, were then performed in order to elucidate the mechanism(s) that trigger the activation of such cells. These studies suggested that reactive microgliosis is prompted by the secretion of mutant SOD1 (Urushitani et al. 2006) , and the aggregated protein was described to be more efficient than the monomeric form in this activity (Roberts et al. 2013) . The relevance of microglia towards the disease manifestations was then tackled in vivo by genetic (Boillee et al. 2006; Gowing et al. 2008) and cell transplantation (Beers et al. 2006) approaches. Thus, reducing the expression of the mutant hSOD1G37R protein selectively in microglial cells was reported not to affect the disease onset, although it slowed down ALS progression in transgenic mice (Boillee et al. 2006). In agreement with these results, transplantation of donor-derived microglia from the spinal cord of hSOD1G93A mice into wild-type animals did not trigger ALS disease (Beers et al. 2006). Yet, transplantation of wild-type microglia induced neuroprotective effects on hSOD1G93A-expressing motor neurons and prolonged the survival of transgenic mice (Beers et al. 2006). Altogether, these studies strengthen the idea that microglial cells modulate ALS progression, rather than influencing the development of the disease .
At the molecular level, the mechanism(s) by which microglia contribute to motor neuron sufferance still needs to be elucidated in details. Yet, a transcriptional profile analysis recently performed on acutely isolated spinal cord microglia from hSOD1G93A transgenic mice revealed that these cells exhibit an ALS-specific phenotype characterized by the concomitant up-regulation of both potentially neurotoxic and neuroprotective factors (Chiu et al. 2013) . Among the molecules presumably mediating microglial toxicity, there are for example reactive oxidant species. In both human and mouse autoptic tissues, the ROS-generating NOX2 enzyme was in fact shown to be up-regulated specifically in microglial cells (Wu et al. 2006) . This event correlated with the appearance of oxidation products and protein carbonylation adducts, including oxidative modifications of insulin-like growth factor 1 (IGF1) receptors, which are critically involved in neuronal survival. The relevance of NOX2 in disease progression was then confirmed by the evidence that genetic ablation of its catalytic subunit ameliorated the phenotype and extended the lifespan of hSOD1G93A ALS mice (Wu et al. 2006). Further insights into the mechanism of NOX2- driven toxicity have been provided a few years later by Harraz and colleagues, who unveiled a new function for the SOD1 enzyme. Along with its dismutase activity, SOD1 was shown to participate to the regulation of NOX2 function by binding to its regulatory protein Rac1. Under reducing conditions, this interaction appears to stabilize Rac1 activation, thus leading to NOX2 activation and superoxide production. However, when the local concentration of oxidant species increases, SOD1 is displaced from Rac1, and NOX2 activity appears to be negatively regulated. Yet, certain mutant forms of SOD1 display a stronger interaction with Rac1, and this leads to both sustained NOX2 activation and excessive superoxide production. Because chronic treatment with the NOX2 inhibitor apocynin tremendously increased the lifespan of hSOD1G93A ALS mice, this mechanism was proposed to contribute to disease progression (Harraz et al. 2008) . The promising outcome of this study subsequently encouraged further investigations on NOX2 inhibitors in the context of ALS. Thus, the neuroprotective properties of diapocynin, a potent NOX2 inhibitor, were addressed in vitro and in vivo (Trumbull et al. 2012) . Despite this drug proved to prevent motor neuron death at lower doses in culture experiments when compared to apocynin, the treatment of hSOD1G93A transgenic mice failed to extend the animal lifespan. Moreover, the dramatic effect on mouse survival originally observed with apocynin could not be replicated in this study (Trumbull et al. 2012) , thus raising serious concerns about the therapeutic potential of these agents. Nonetheless, the pathway(s) leading to NOX2 activation, and their contribution to ALS pathogenesis, remain of great interest for the scientific community. In fact, the enzyme protein disulfide isomerase, which assists the oxidative protein folding in the ER, was recently shown to be up-regulated in microglia from hSOD1G93A mice. This suggests that the unfolded protein response (UPR) does not occur only in ALS motor neurons, but also in microglial cells (Jaronen et al. 2013) . Importantly, UPR was reported to trigger the activation of microglial NOX2 and, thus, to increase the production of superoxide in culture experiments (Jaronen et al. 2013). These findings suggest that UPR, initiated by protein misfolding, may lead to NOX2 activation and ROS generation. However, recent studies propose alternative pathways leading to the activation of NADPH oxidase. For example, experiments in vitro showed that stimulation of the purinergic receptor P2 × 7 in mutant hSOD1G93A-expressing microglia results in enhanced NOX2 activity and ROS production, thus suggesting a deleterious role for the P2 × 7 receptor in ALS (Apolloni et al. 2013b) . At variance with these results, genetic ablation of the P2 × 7 receptor in hSOD1G93A mice, however, exacerbated gliosis and motor neuron cell death; anticipated the clinical onset of the disease; and worsened ALS progression (Apolloni et al. 2013a). The protective effect of P2 × 7 suggested by these results in vivo was completely unexpected, given that stimulation of the P2 × 7 receptor had been linked also to the induction of a neurotoxic phenotype in hSOD1G93A astrocytes (Gandelman et al. 2010) and to motor neuron apoptosis (Gandelman et al. 2013). Altogether, these observations reveal that the P2 × 7 receptor exerts a complex dual role in ALS. Evidence in favor of a neuroprotective role for microglia in the context of ALS was provided by a recent study addressing the biological significance of the glycosaminoglycan keratan sulfate (KS) (Hirano et al. 2013) . In both autoptic ALS tissues and hSOD1G93Atransgenic mice, KS was found to be highly expressed by a specific subpopulation of microglial cells exerting anti-inflammatory functions during the early phase of the disease. Noteworthy, genetic ablation of KS in the CNS of hSOD1G93A mice resulted in marked reduction of anti-inflammatory microglia, and this correlated with acceleration of the clinical symptoms and shortening of the animal lifespan (Hirano et al. 2013) .
More recently, microglia was also describe to dialogue with the peripheral immunocompetent cells, an interaction that proved to be both beneficial and deleterious (reviewed in Appel et al. 2010) . Several lines of evidence indicate that T cells infiltrate the spinal cord of ALS patients (Troost et al. 1989; Kawamata et al. 1992; Engelhardt et al. 1993) and mutant SOD1 transgenic mice (Beers et al. 2008; Chiu et al. 2008) . Of note, infiltrating T lymphocytes were described to slow down disease progression in mice by modulating the microglial inflammatory response (Beers et al. 2008, 2011; Chiu et al. 2008; Henkel et al. 2013) . Yet, spinal cord microglia were also reported to recruit peripheral monocytes to the CNS, a process that critically impairs neuronal viability and the mouse survival (Butovsky et al. 2012) .
In conclusion, these findings globally suggest that the neuroinflammatory response driven by microglia includes both neuroprotective and neurotoxic aspects in ALS. Therefore, the possibility of establishing a successful therapeutic effect relies on the selective targeting of the toxic components of the immunoreaction, rather than on the non-specific suppression of the immunocompetent response .
11.5 Oligodendrocytes and Other Glial Cell Types
While astrocytes and microglia have been at the center of the investigations on ALS for almost a decade, other glial cell populations have attracted the attention in more recent times. Among others, these latter include oligodendrocytes and Schwann cells , the myelin-forming cells of the central and peripheral nervous system, respectively. Although early analyses of mutant SOD1 transgenic mice revealed SOD1-immunopositive inclusions in oligodendrocytes (Stieber et al. 2000) , this observation was not pursued immediately thereafter, and these cells have been involved in ALS pathogenesis quite recently. In both ALS patients and mutant hSOD1G93A transgenic mice, it has been consistently reported a loss of the monocarboxylate transporter 1 (MCT1), a protein that is highly expressed in oligodendrocytes, and which is deputed to provide motor neurons with the metabolic substrate lactate (Lee et al. 2012; Philips et al. 2013) . Noteworthy, ubiquitous genetic ablation of MCT1 in mice was described to cause axonopathy (Lee et al. 2012). Furthermore, selective reduction of its expression in oligodendrocytes resulted in axonal sufferance (Lee et al. 2012) . This suggests that the shuttling of lactate from oligodendrocytes, through MCT1, is crucial for the energy supply to axons, and any disturbance of this transport can lead to axon dysfunction and, eventually, to neuronal cell death.
More recently, oligodendrocytes themselves were described to be a direct target of the disease. An apoptotic oligodendrocytic phenotype was, in fact, detected in the ventral grey matter of the spinal cord from mutant hSOD1G93A transgenic mice before actual motor neuron loss became evident. Surprisingly, the overall number of oligodendrocytes was fully preserved (Philips et al. 2013). In demyelinating diseases, mature oligodendrocytes are typically replaced by the differentiation of NG2+ cells , which behave as precursors committed to the oligodendrocyte lineage (Chang et al. 2000) . In keeping with this, investigations of the NG2+ cell fate in symptomatic mutant SOD1 mice revealed that they exhibit an increased proliferation rate (Magnus et al. 2008; Kang et al. 2010, 2013; Philips et al. 2013) . The occurrence of this event is accompanied by a more frequent differentiation of NG2+ cells into oligodendrocytes (Magnus et al. 2008; Kang et al. 2010, 2013; Philips et al. 2013). Yet, at the time when mice show overt signs of the disease, the concomitant degeneration of early-born oligodendrocytes and, thus, the accelerated turnover of these cells, result in grey matter demyelination in ALS mice and human CNS (Kang et al. 2013). In addition, newly generated oligodendrocytes display reduced MCT1 expression, and thus fail to provide motor neuron with metabolic support (Kang et al. 2013; Philips et al. 2013). Since oligodendrocyte myelination is regulated by lactate (Rinholm et al. 2011) , it was proposed that reduced levels of MCT1 in oligodendrocytes may affect the process of myelination, in addition to depriving motor neurons of critical metabolic substrates (Magnus et al. 2008; Kang et al. 2010, 2013; Philips et al. 2013). A role for NG2+ cells and their oligodendrocyte progeny in ALS development was corroborated also by gene targeting experiments. Thus, diminishing mutant SOD1 expression in these cells was shown to delay the onset of the disease and to extend the life span of mutant hSOD1G37R transgenic mice (Kang et al. 2013).
In the peripheral nervous system (PNS), individual axons are myelinated by the Schwann cells , the major glial component of the PNS. An initial indication of the involvement of this glial cell population in the pathogenesis of ALS was provided by the early observation that femoral nerves from post-mortem cases displayed a certain degree of myelin disruption (Perrie et al. 1993) . Later studies in mutant hSOD1G93A mice provided further insights into the role of these cells in ALS by revealing that they exhibit signs of distress at the asymptomatic stage (Keller et al. 2009) . Yet, investigations addressing the impact of Schwann cells on ALS pathogenesis in vivo by cell-specific expression or ablation of mutant SOD1 provided slightly divergent outcomes. Thus, transgene-driven expression of mutant hSOD1G93A protein in Schwann cells resulted neither detrimental for motor neurons nor deleterious to the disease manifestations (Turner et al. 2010) . On the other hand, removal of mutant hSOD1G37R from Schwann cells by gene excision experiments accelerated disease progression in vivo (Lobsiger et al. 2009) . Considering the intimate relationship between motor neuronal axons and myelin, these results indicate that Schwann cells certainly deserve further attention in order to definitely elucidate their contribution to the disease.
Related posts:
 Experimental Treatment of Acquired and Inherited Neuropathies
Possible Therapeutic Targets in Microglia
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
Enteric Glial Cells: Implications in Gut Pathology
Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Experimental Treatment of Acquired and Inherited Neuropathies
Possible Therapeutic Targets in Microglia
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
Enteric Glial Cells: Implications in Gut Pathology
Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


