Introduction
Epilepsy is one of the most common neurological disorders. The need to accurately evaluate and treat seizures is within the realm of many physicians, including neurologists, internists, family doctors, paediatricians, geriatricians, emergency room physicians and obstetricians just to name a few. Seizures can present as a symptom in outpatient clinics as well as in hospitalized patients. Epilepsy affects a broad age group, has a diverse spectrum of aetiologies, and has a variable response to treatment. As one can imagine, making a diagnosis of epilepsy poses several challenges. The diagnosis is made with the highest degree of confidence when seizures are recorded during a video-electroencephalography (
EEG) evaluation. For most patients who present with a history of seizures, results from video-EEG testing is usually not available and so the diagnosis is made using other available information. A careful and comprehensive history often enables an accurate diagnosis of epilepsy or may suggest the need for further testing, as well as helping to guide treatment decisions. The historical information comes from both the patient as well as from observers who have seen a patient’s seizures. Whereas many additional tests such as neuroimaging and
EEG may well be indicated in the evaluation of epilepsy, the importance of history cannot be overemphasized. Throughout this chapter and subsequent chapters in this atlas, various aspects of epilepsy will be highlighted. The intent of this atlas is for physicians to use it as a resource for a quick and easy guide for the assessment and diagnosis of various forms of epilepsy. There are some brief references in regards to the treatment of epilepsy, but this is not the main intent of this atlas.
Epidemiology
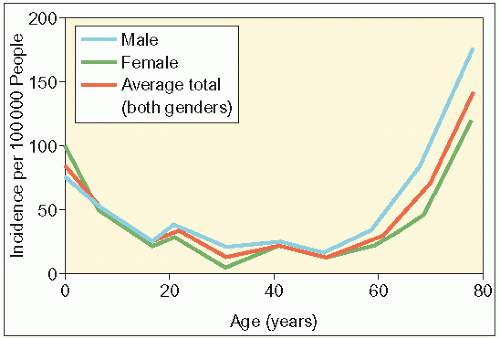
Every year 20 people in 100 000 will have had a seizure. For the majority, it will be their first and only seizure. A diagnosis of epilepsy can only be made when there has been at least two unprovoked seizures or recurring seizures with more than a 24-hour interval between each seizure. Epilepsy is a disease that knows no geographical, racial or social borders. The disorder affects the sexes equally with an approximate prevalence of 7.1 per 1000 people. The annual incidence has been reported to be 48 per 100 000 people with a higher incidence noted in developing countries, possibly due to poorer nutrition, hygiene and perinatal care, as well as an increased risk of cerebral infection.
The incidence of epilepsy peaks in infancy as well as in individuals over 60 years of age. Approximately 1% of the US population meets the diagnostic criteria for epilepsy but the actual prevalence may be significantly higher. Within 5 years of onset of seizures, 50-60% of patients will have entered a long period of remission. Up to 30% of patients will develop medically refractory epilepsy, despite multiple anticonvulsant medication trials. The standardized mortality rate associated with epilepsy is increased two to three times, with the excess mortality often directly linked to seizures. Compared with the general population, patients with epilepsy are more likely to be involved in accidents and suicides. Patients with epilepsy are also at greater risk of sudden unexpected death; it is estimated that there is one death per 250 people with severe and refractory epilepsy.
Seizure types
The classification of epilepsies is a controversial subject. There are many classification systems including the International League Against Epilepsy (
ILEA) classification (described further on
pp. 11-13). Currently the classification is undergoing some revision. In obtaining an historical account of the seizure it is important to obtain the description from both the patients and observers. The patient’s account will allow a description of various aura and symptoms prior to the loss of awareness or consciousness if they occur. The observer’s description will give information during periods where the patient has no memory of the seizure. The descriptions are grouped under major headings based on various classification systems. The seizure classification described below includes terms being used currently in the ILAE classification as well as terms being used in the semiological classification (which has been advocated by some epilepsy centres around the world). The designation ‘ILAE’ for the ILAE classification or ‘SE’ for the semiological classification will denote terms specific to each classification system. One of the main disadvantages of the ILAE is that it is based not only on historical information but makes assumptions regarding the associated underlying epilepsy producing the seizure type, including on
EEG data that may not be available (see below).
Aura
Auras occur at the beginning of a seizure and are only appreciated by the patient and are subjective symptoms. They usually are very brief and can occur in isolation. They are not detectable to the observer. Some examples of auras include: somatosensory auras (including tingling or alteration in sensation in a somatotopic distribution of the body); visual or auditory auras (can consist of hallucinations and illusions);
autonomic auras (can manifest with symptoms of tachycardia or goose bumps); abdominal auras (typically described as a rising sensation from the abdomen to the thorax or throat along with complaints of nausea); psychic auras (are complex and described as experiential symptoms of ‘déjà vu’, fear, or ‘jamais vu’). The patient may exhibit some behavioural response during their aura, such as a fearful expression, which is simply a reaction to either the symptom of the aura or knowing that they are about to experience a seizure.
Dialeptic seizure (SE)
This term is used when there is an alteration of either consciousness or awareness as the major clinical manifestation of the seizure. Typically, the patient has no recollection of the seizure and their other associated motor manifestations. An absence seizure (ILAE) not only has an alteration of consciousness but also requires that whatever electroclinical information is available suggests that the seizure arose from generalized epilepsy.
Motor seizure (SE)
This terminology is used for seizures that have some significant motor component.
Simple motor seizures (SE) are those that comprise involuntary movements that bear no resemblance to physiological or natural movements. These movements can be elicited through stimulation of the primary motor cortex (Brodmann’s area 4 and 6) or the supplementary sensorimotor area (Brodmann’s area 6). At the beginning of these seizures consciousness may be preserved; however, it may be lost as the seizure develops.
In complex motor seizures (SE) they comprise complex movements that appear physiological, such as bicycling or moving about. However, these movements are inadequate or inappropriate for the patient’s setting. During these seizures awareness is usually, although not always, altered.
Clonic seizures (SE and ILAE—although a prefix term ‘generalized’ is typically used in the ILAE classification)
These seizures consist of periodic, repetitive, short contractions of various muscle groups. Often these movements are found in the distal extremities, face or tongue. Epileptic discharges from the primary motor region (area 4) or the premotor region (area 6) lead to these movements. In the semiological classification clonic seizures may involve the entire body, when it is called generalized, or may be more limited, such as involving only the left arm, in which case it would be termed left arm clonic seizure. This also holds true for tonic seizures.
Tonic seizure (SE and ILAE—although a prefix term ‘generalized’ is typically used in the ILAE classification)
These seizures comprise a persistent contraction of one or more muscle groups, which lead to tonic posturing. They most often appear in the proximal musculature and have a preference for the contralateral side to the epileptic focus. Epileptic stimulation of the supplementary sensorimotor region (medial area 6) leads to these movements.
Generalized tonic-clonic seizure (SE and ILAE)
This seizure type describes what has become known in the vernacular as ‘grand-mal seizure’. It denotes a generalized seizure with semiology combining a generalized clonic seizure with a generalized tonic seizure.
Generalized atonic seizure (SE and ILAE)
This seizure type also denotes a seizure in which all motor tone is lost and the patient drops to the floor. Patients with these seizures are more prone to serious head injuries.
Myoclonic seizures (SE and ILAE—although a prefix term ‘generalized’ is typically used in the ILAE classification)
These seizures comprise of isolated, quick twitches, which can be generalized or focal in nature.
Versive seizures (SE)
These seizures consist of a forced unnatural turning of the heads and or eyes in one direction. The movement may be a smooth tonic contraction or be clonic. The deviation of the head and/or eyes may sometimes be followed by turning the trunk in the same direction. The important feature of this seizure type is that the movement leads to an unnatural, tonic posturing, which helps differentiate it from other nonversive head movements.
Complex partial seizure (ILAE)
A seizure in which consciousness is impaired, and electroclinical information suggests that the seizure arises from a focal epilepsy.
Hypomotor seizure or behavioural arrests
Patients during these seizures demonstrate reduced motor activity, although certain complex motor actions are still
possible such as sitting upright or a blank stare. However, consciousness cannot be tested in this group of patients either because of mental disability or because of their young age.
Automotor seizures or complex partial seizures
Automatisms are involuntary, organized movements, such as mictition, swallowing or nose rubbing. In the setting of these seizures the automatisms appear inappropriate to the patient’s setting.
Hypermotor seizures or complex partial seizures
These seizures are characterized by a series of complex motor movements in the proximal extremities that appear fervid and sometimes even violent or bizarre. Automatisms are not observed in these seizures.
Risk factors
Complex febrile seizures
These seizures last more than 15 minutes with a predominantly unilateral symptomatology that recur within a single infection. They are usually associated with a rapid increase in temperature due to a viral illness. Approximately one-third of all febrile seizures are complex, usually occurring very early in the infection and may be the first clinical manifestation. They can, however, have a genetic basis and genetic studies have shown an autosomal dominant inheritance pattern of the SCN1A gene in families with generalized epilepsy and febrile seizures. Two to 10% of children with a history of febrile seizures will develop epilepsy later in life, in comparison with 0.5% of children with no history. It should be noted, however, that the risk for development of epilepsy is greater in children with a pre-existing neurodevelopmental dysfunction and the febrile seizure may be the first clue to their predisposition to epilepsy.
Head trauma
Injuries of this type can be divided into open and closed head trauma. Post-traumatic epilepsy occurs much more frequently with open head trauma, and the risk is greatest if damage occurs in large areas or involves frontal and temporal lobes, with 50-60% of patients experiencing their first (late) within the first 12 months after injury. Dural breach, encephalomalacia, intracranial haematoma and long post-traumatic amnesia have all been found to increase the risk of developing epilepsy.
Closed head trauma can also increase the risk for developing epilepsy; however, mild injury is not associated with any increased risk. One to 4% of patients with moderate injury, defined as head injury complicated by skull fracture or more than 30 minutes of post-traumatic amnesia, will develop epilepsy. As many as 10-15% of patients with severe head trauma—head injury with post-traumatic amnesia longer than 24 hours, cerebral contusion or intracranial haematoma—will develop epilepsy.
Family history
It is important to identify other family members who have been diagnosed with epilepsy because an increased prevalence has been established in affected families, which is consistent with both common environmental exposures as well as polygenic or multifactorial genetics. Cohort studies have shown there to be a two- to threefold increased risk of developing epilepsy in siblings and children of epileptics. The genetics of epilepsy will be discussed later.
Developmental delay
There has been an association between epilepsy and developmental delay, and careful history taking might hint towards an epileptic aetiology. In a paediatric study developmental delay or mental retardation was present in 37% of patients. Many of the aetiologies, such as cortical dysplasia, associated with paediatric epilepsy are also associated with cognitive impairment of varying degrees.
Some enzyme deficiency diseases may also lead to a developmental delay and be associated with epilepsy. These diseases often have a genetic basis and the exact mutation is known, which results in a protein dysfunction or deficiency, thus stunting cognitive development. Examples include Gaucher disease, Niemann-Pick disease type C, various lysosomal and peroxisomal disorders, porphyria, pyridoxone deficiency and Wilson disease.
Central nervous system infections
Central nervous system tumours
Comorbidities
Epilepsy is associated with several comorbidities that impair patient health and the proper management of a patient with epilepsy includes treating these as well as gaining control over seizures. Approximately 5% of all annual visits to the emergency department are related to injuries as a result of seizures, such as head trauma, burns, falls and lacerations. Most other common comorbidities fall under three broad headings: psychiatric, cognitive impairment and endocrine dysfunction. There is a much higher rate among epileptics in comparison with the general population of anxiety disorders (19-66%) and depression (20-57%). These disorders probably have several aetiologies and are related to the disease pathology itself, the sedative effects of antiepileptic drugs (
AEDs), as well as the psychosocial stigma appointed to seizures. Cognitive impairment has already been dealt with under ‘Developmental delay’. Female patients with epilepsy are more likely to experience reproductive endocrine dysfunction than are female patients without epilepsy. Ovulatory dysfunction and hyperandrogenism in the absence of thyroid or adrenal disease usually takes the form of polycystic syndrome and is a common comorbidity among female patients. Other reproductive difficulties may arise from treatment with certain
AEDs, which can reduce sexual drive and have teratogenic properties on the fetus. Epileptics are more likely to suffer from insulin resistance, diabetes mellitus type 2, hypertension, dyslipidaemia and cardiovascular disease; the exact pathophysiology has not been determined but probably involves neuroendocrine effects of seizures and/or
AEDs.