Fig. 1
Overview of adult hippocampal neurogenesis: summary of five developmental stages during adult hippocampal neurogenesis. 1 Activation of quiescent radial glia-like cell in the subgranular zone (SGZ). 2 Proliferation of non-radial precursor and intermediate progenitors. 3 Generation of neuroblasts. 4 Integration of immature neurons. 5 Maturation of adult-born neurons. Expression of stage-specific markers, sequential process of synaptic integration, and critical periods regulating survival and plasticity are shown. Dentate gyrus (DG), molecular layer (ML), granule cell layer (GCL), subgranular zone (SGZ), glial fibrillary acidic protein (GFAP), brain lipid binding protein (BLMP), doublecortin (DCX), neuronal nuclei (NeuN), gamma-aminobutyric acid (GABA), and long-term potentiation (LTP). Reprinted from Neuron, 70-4, Ming and Song (2011), with permission from Elsevier
Newborn neurons undergo morphological and physiological maturation by extending axons into the hippocampal CA3 region and dendrites into the molecular layer of the dentate gyrus (Stanfield and Trice 1988; Gu et al. 2012). Inputs from the entorhinal cortex stimulate formation of spines and functional synapses (Markakis and Gage 1999; Zhao et al. 2006). Immature neurons are at first tonically depolarized by gamma-aminobutyric acid (GABA) released from interneurons. Depolarizing GABAergic input was found to increase proliferation and differentiation in the neuronal precursors (Tozuka et al. 2005; Ge et al. 2007a). A final step in neurogenesis is reception of synaptic glutamate input via AMPA and NMDA receptors, which mediate depolarization and promote survival of newborn neurons (Tashiro et al. 2006). Young neurons are hyperexcitable, which is thought to enhance their participation in synaptic plasticity . A portion of the new neurons will be integrated into the hippocampal network over a time span of 2 months, during which axosomatic, axodentritic, and axospinous synaptic contacts are established (van Praag et al. 2002; Toni et al. 2007). Survival is experience and input dependent and over 50 % of new cells die by apoptosis, many within the first few days after exiting the cell cycle (Dayer et al. 2003; Tashiro et al. 2007; Sierra et al. 2010).
Most studies have used immunocytochemical detection of BrdU as a cell proliferation marker (Taupin 2007). After BrdU injection, brains are collected for processing at a time determined by the research question. To identify newly proliferating cells, brains are typically harvested 2 h after BrdU injection (approximate duration of BrdU bioavailability), which captures cells in the S-phase (~7–9 h in rats) before entering mitotic cell division (Figs. 2a, 5a). If cell survival is of interest, animals are kept alive for days to weeks after BrdU injection (Figs. 2b, 5b). The population of cells labeled by a single injection of BrdU doubles over about 2 days, and may expand for up to 4 days (Dayer et al. 2003). Therefore, to measure the effects of a procedure (e.g., sleep deprivation) on survival, independent of proliferation, BrdU must be administered 4 or more days prior to the procedure. Endogenous mitotic cell cycle stage-specific proteins can also be immunolabeled to identify proliferating cells. For example, Ki67 (Fig. 2c), a nuclear protein, is expressed during all phases of the cell cycle except the resting phase (G0) and early G1 (Kee et al. 2002). Proliferating cell nuclear antigen (PCNA ), an auxiliary protein of the DNA polymerase δ occurs in G1–G2 with peak expression at the G1/S interphase and phosphorylated histone H3 is restricted to the G2 and M-phase of the cell cycle (Galand and Degraef 1989; Hendzel et al. 1997). Such endogenous proliferation markers can be used to confirm that differences in BrdU labeling observed following a procedure are not due to changes in the bioavailability or distribution of exogenous BrdU. To study cell differentiation and maturation of particular phenotypes, newborn cells can be labeled or double-labeled with cell-type specific markers. Commonly used markers include DCX , NeuroD, and TuJ1 for immature neurons, NeuN for mature neurons, and GFAP or calcium-binding proteins S100, S100β for glial cells (Abrous et al. 2005). See Fig. 3.
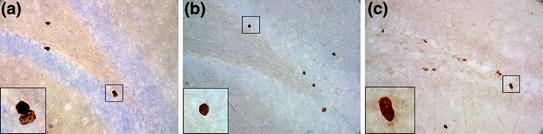
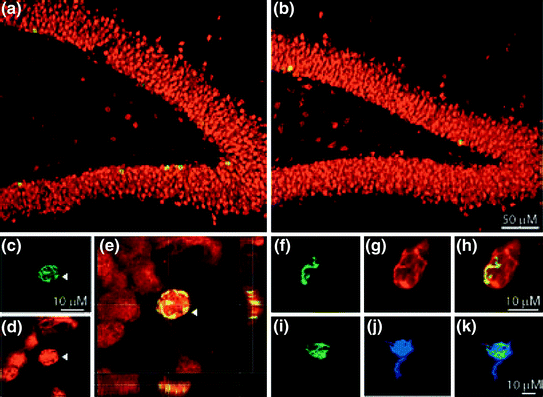
Fig. 2
Representative examples of immunohistochemical analysis for cell proliferation and cell survival in the adult dentate gyrus. Peroxidase staining with DAB as reporter group. 400× magnification, inserts 1000×. a Cell proliferation of BrdU+ cells, 2 h survival. b BrdU+ cell survival (3 weeks). c Ki67+ cell proliferation
Fig. 3
Confocal images of fluorescence immunolabeling in the dentate gyrus of control (a) and sleep deprived (b) rats. a–e Double labeling with BrdU (c green) and NeuN (d red), a marker for new neurons. e Merged picture of a BrdU+ and NeuN+ cell. f–h Co-localization of BrdU (f green) with DCX (g red). i–k Co-localization of BrdU (i green) and S100β (j blue), a marker for astrocytes. Reprinted from EJN, Guzman-Marin et al. (2005), used with permission from Wiley and Sons
4 Regulation and Function of Adult Hippocampal Neurogenesis
Young adult rats produce about 9,000 new cells in the dentate gyrus per day (Cameron and McKay 2001) but the ultimate disposition of new cells is influenced by a wide range of physiological, pathological, and behaviorally mediated stimuli affecting the level of cell proliferation, differentiation, maturation, and survival (Ming and Song 2005). The role of adult-born granule cells in hippocampal function is still under investigation (Song et al. 2012). Participation in learning and memory processes is suggested by the electrophysiological properties of new neurons. Young neurons show robust long-term potentiation (LTP) with a low threshold for LTP induction (Schmidt-Hieber et al. 2004; Ge et al. 2007b). This results in a preferential recruitment of new neurons by behavioral activation (Ramirez-Amaya et al. 2006; Kee et al. 2007). Elimination of hippocampal neurogenesis by irradiation has been shown to severely reduce induction of LTP (Snyder et al. 2001).
Direct tests of a role for new neurons in learning and memory have yielded mixed results. For example, antimitotic drugs can eliminate certain forms of hippocampal-dependent learning, such as trace eye blink conditioning and trace fear conditioning, but not others, such as contextual fear conditioning or spatial navigation learning (Shors et al. 2001, 2002). Nonetheless, a picture is now emerging that adult hippocampal neurogenesis is involved in spatial and temporal pattern separation, trace conditioning, contextual fear conditioning, long-term memory retention, and clearance of hippocampal memory traces (Koehl and Abrous 2011). Importantly, the relationship between adult neurogenesis and learning appears to be reciprocal, because training on these tasks can stimulate cell proliferation or survival (Gould et al. 1999a), as does exposure to enriched environments (Kempermann et al. 1997). An increased pool of new neurons is hypothesized to enhance the capacity for behavioral plasticity, and facilitate adaptation to environmental novelty and complexity (Kempermann 2008).
Hippocampal neurogenesis has also been implicated in the regulation of mood. The hippocampus has reciprocal connections with the amygdala and the prefrontal cortex, and plays a central role in emotions. Stress can precipitate or exacerbate mood disorders, and both major depression and stress are associated with impaired hippocampal function and morphological changes. Negative regulation of neurogenesis by stress (Cameron and Gould 1994; Mirescu and Gould 2006; Hanson et al. 2011), and positive regulation by antidepressant treatments [e.g., SSRIs and exercise; van Praag et al. (1999), Malberg et al. (2000), Leuner et al. (2006), Sahay and Hen (2007)] with a time course mimicking the onset of therapeutic efficacy, suggested a hypothesis that neurogenesis may play a major role in the pathophysiology and/or recovery from major depression (Jacobs et al. 2000). This hypothesis continues to be explored (Sahay and Hen 2007; Eisch and Petrik 2012; Patricio et al. 2013).
Sleep, like neurogenesis, is implicated in the regulation of memory and mood, and many of the factors that regulate neurogenesis also affect sleep or are affected by sleep. This raises the possibility that sleep may regulate adult neurogenesis, either directly or indirectly, as follows:
1.
Sleep may represent a physiological state required for neurogenesis that gates or facilitates critical molecular events in one or more of the stages from cell proliferation to functional neurons. In this relationship, specific events in neurogenesis would occur only during sleep. Sleep would, therefore, be necessary for neurogenesis, and neurogenesis would represent a function of sleep.
2.
Sleep may be necessary for maintaining optimal functioning of neural and endocrine processes that in turn regulate neurogenesis. In this relationship, specific events in neurogenesis would not be limited to sleep, but some amount of sleep would be required for neurogenesis to proceed optimally.
3.
Sleep disruptions may evoke nonspecific responses such as stress responses or behavioral changes that reduce neurogenesis. In this relationship, sleep would play no special role in neurogenesis, but manipulations of sleep could nonetheless affect the process.
In the following sections, a role for sleep in the regulation of neurogenesis is considered, based on evidence from correlation, deprivation, and stimulation studies.
5 Sleep and Neurogenesis: Correlational Studies
If sleep regulates neurogenesis, then variations in sleep may be paralleled by variations in neurogenesis. In mammals, natural variations in sleep occur across the day (nocturnal and diurnal sleep chronotypes), across the lifespan, and in some species across seasons. One or more processes in neurogenesis could, therefore, vary with time of day, time of year, or time of life.
5.1 Seasonal and Lifespan Correlations
There is a robust correlation between sleep and neurogenesis across the lifespan . Cell proliferation and neurogenesis are maximal during early development when daily sleep amounts are greatest (Alfoldi et al. 1990). The steepest decline in the production of new neurons occurs in the early life stages, likely reflecting a transition of neural stem cells to a quiescent state, resulting in a depletion of the pool of proliferating cells (Lugert et al. 2010; Kempermann 2011b). Cell proliferation in rodents further declines with age (Heine et al. 2004), although this has not yet been related specifically to changes in sleep parameters.
Seasonal variability in sleep expression occurs in some mammals that migrate or hibernate, but this has not been specifically related to rates of neurogenesis. Neurogenesis in adult birds is beyond the scope of this chapter, but it is worth noting that annual rhythms of neurogenesis and sleep are evident in various avian species. Some species of songbirds each year regrow brain structures necessary for song learning (Barnea and Pravosudov 2011); whether this is associated with variations in sleep has not been directly assessed.
5.2 Correlations with Time of Day
In most mammals, sleep exhibits a prominent daily rhythm driven and synchronized to the solar day by a master retinorecipient circadian clock in the hypothalamic suprachiasmatic nucleus (SCN).
The cell cycle of proliferating cells in the adult rat hippocampus is estimated to be ~24–25 h in duration (Cameron and McKay 2001). If the cell cycle is gated by circadian or sleep–wake dependent factors, then a daily rhythm of cell proliferation should be detectable by comparing BrdU labeling following administration at different times of day. Note that BrdU would be expected to label cells entering the cell cycle beginning 7–9 h (the duration of the S-phase) before its administration. Evidence for a daily rhythm has been obtained using this approach in adult male rats, with four injection times and 2 h post-injection survival time (Guzman-Marin et al. 2007b). BrdU labeling in the granule cell layer/subgranular zone was found to be maximal late in the light (rest) period , with double the number of labeled cells observed when BrdU was administered 9 h after lights-on compared to the beginning of the light period and early or late in the dark period .
Enhanced proliferation near the end of the sleep period is consistent with a neurogenic role for sleep, but other studies have observed either a differently phased rhythm, or no time of day effect (Fig. 4). One study that also used BrdU injections with a 2 h survival time reported 22 % more labeled cells following injections at the end of the dark period relative to the end of the light period (Junek et al. 2010). Another study using the endogenous proliferation marker Ki67 reported a significant rhythm, but with maximum proliferation rates in the middle of the dark period, 6 h after lights-off, and lowest rates in the middle of the light period (Gilhooley et al. 2011). Two studies found no daily variation. In one case, proliferation was measured in single-housed rats using BrdU injections at hours 0 or 9 of the light period, with a 2 h post-injection survival (Mueller et al. 2011). In a second study, BrdU was administered at one of 4-time points but with a 24 h post-injection survival time (Ambrogini et al. 2002). There was no significant effect of time of day, although the longer survival time would likely attenuate time of day differences, depending on how long BrdU remains available, and whether this varies with time of day.
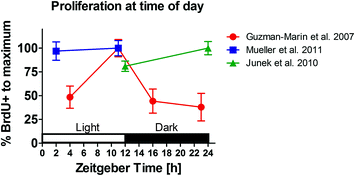
Fig. 4
Circadian variation of cell proliferation in male Sprague–Dawley rats. Replotted data from Guzman-Marin et al. (2007a, b), Mueller et al. (2011) and Junek et al. (2010). Animals were kept in a standard 12:12 h light/dark cycle. Cell proliferation was measures with BrdU and 2 h survival time. X-axis: time of perfusion at Zeitgeber time (ZT, with ZT0 representing light on, as by convention). Guzman-Marin and colleagues reported a circadian variation of cell proliferation with peak levels at the end of the light period. However, the topic remains inconclusive. Neither Mueller et al. (2011) nor Junek et al. (2010) were able to confirm a daily rhythm of adult hippocampal cell proliferation with peak levels of the S-phase at the end of the light period
The cell cycle of proliferating cells in the adult hippocampus of C57BL/6J and BALB/cByJ mice has been estimated to be ~12–14 h (Hayes and Nowakowski 2002) and thus could complete 2 cycles per day, unless progression is stopped in G0, pending appropriate signals from the circadian clock or sleep-wake states. Three studies reported no daily rhythm of cell proliferation in the granule cell layer/subgranular zone assessed using BrdU or Ki67 in mice housed under sedentary conditions, e.g., without a running wheel (Holmes et al. 2004; Kochman et al. 2006; van der Borght et al. 2006). A fourth study also reported no daily rhythm of BrdU labeling (S-phase), but did find evidence for a daily rhythm of labeling using antibodies to phosphohistone H3 (a marker for M-phase) and phospho cdc2 (marking transition from G2 to M-phase) (Tamai et al. 2008). The authors proposed a model whereby progenitor cells enter the cell cycle at any time of day, but progress through the M-phase primarily at night, producing a nocturnal peak of cell proliferation.
A single study of cell proliferation in the dentate gyrus of the Syrian hamster reported no difference in BrdU labeling following injections at the end of the light period compared to the end of the dark period (Smith et al. 2010). This study used 24 h survival times following injections, which may not be optimal for detecting daily rhythms. Surprisingly, a time of day effect was observed when survival time was extended to 72 h; by contrast with the 24 h survival group, the number of labeled cells in the 72 h survival group was approximately doubled in hamsters that received BrdU at the end of the light period, whereas no increase was seen in hamsters that received BrdU at the end of the dark period. This result implies that cells in the S-phase of mitosis near the end of the light period continue to divide, expanding the number of labeled cells, while those labeled near the end of dark period do not continue to divide, or have reduced survival.
Two studies have reported a significant daily rhythm of BrdU labeling in the hilus, in mice (Kochman et al. 2006) and rats (Guzman-Marin et al. 2007b). In both cases, the peak of labeling was observed following BrdU injections late in the light period. The hilus gives rise primarily to glia cells, thus these results suggest a daily rhythm of hippocampal gliogenesis. Two other rat studies did not detect a rhythm of BrdU labeling in the hilus (Ambrogini et al. 2002; Mueller et al. 2011).
5.3 Daily Rhythms of Cell Proliferation: Effects of Wheel Running
Wheel running stimulates cell proliferation and neurogenesis (Vivar et al. 2012). Given that rats and mice are primarily nocturnal runners, free-access to running wheels might be expected to induce a daily rhythm in cell proliferation. A few studies have examined this, again with mixed results. In one study, cell proliferation, cell survival, and the total number of new neurons where all significantly increased relative to sedentary control mice when a running wheel was provided each day for 1 or 3 h at the beginning or in the middle of the dark period, but not when provided in the middle of the light period (Holmes et al. 2004). The mice ran in the wheels at all three times, but more at night, therefore the time of day differences would be consistent with the known effect of exercise on neurogenesis. In that study no daily rhythm of cell proliferation was evident in the sedentary control groups, suggesting that an effect of time of day in mice is likely to be related to waking activity rather than to sleep. A similar result was found using Ki67 as a proliferation marker, with more labeled cells evident near the end of the dark period in mice provided with a running wheel, but not in sedentary controls (van der Borght et al. 2006). One other study observed evidence for a nocturnal peak of proliferation, using a marker for M-phase, but, only in sedentary mice, and not in mice with free-access to a running wheel (Tamai et al. 2008).
5.4 Summary: Is There a Daily Rhythm of Cell Proliferation in the Hippocampus?
The available evidence provides no consensus. Of the five rat studies available, two found a rhythm with a nighttime peak, one found a rhythm with a daytime peak, and two found no rhythm. The discrepancy between the Guzman-Marin et al. (2007b) and Mueller et al. (2011) results was unexpected, given that the latter study was modeled closely after the former. A methodological factor that did vary between the two studies was the use of group versus single housing, respectively. Housing conditions have been shown to modulate stress and exercise effects on hippocampal neurogenesis, and possible modulation of time of day effects as well cannot be ruled out (Stranahan et al. 2006; Kannangara et al. 2009; Leasure and Decker 2009). Of the four mouse studies available, one found evidence for a rhythm with a nighttime peak (Tamai et al. 2008), and three found no rhythm when mice were housed under sedentary conditions (Holmes et al. 2004; Kochman et al. 2006; van der Borght et al. 2006). Three of these studies included a running wheel condition, and, remarkably, the pattern of results across studies was inverted; the one study that reported a daily rhythm under sedentary conditions found no rhythm when a wheel was available (Tamai et al. 2008), while two that reported no rhythm under sedentary conditions obtained evidence consistent with a nighttime peak associated with nocturnal running (Holmes et al. 2004; van der Borght et al. 2006). Finally, four studies have examined time of day effects on cell proliferation in the hilus, with positive evidence in one mouse study (Kochman et al. 2006) and one rat study (Guzman-Marin et al. 2007b), and negative evidence in two other rat studies (Ambrogini et al. 2002; Mueller et al. 2011). The diversity of results does not appear to reflect a species difference, and is more likely to be related to the use of different proliferation markers, injection and survival protocols, and housing conditions. Although consensus is lacking, the results overall are more consistent with regulation of cell proliferation by waking activity rather than by sleep. Ultimately, correlations are silent about causality and must be interpreted in the light of experimental studies employing stimulation and deprivation methods.
6 Sleep and Neurogenesis: Deprivation Studies
If sleep regulates neurogenesis, then sleep disruption should alter the process at one or more levels. Several laboratories have addressed this issue over the last decade. A range of sleep deprivation durations have been evaluated, from 6 h to a week. Most experiments have targeted total sleep deprivation or REM sleep deprivation, but sleep restriction and sleep fragmentation have also been examined. Methods used include gentle handling , exposure to novel objects , forced locomotion , and confinement to a small platform-over water . All of these methods have potential confounds, related to stress and activity levels, which can affect neurogenesis independently of sleep deprivation. Therefore, appropriate control groups are essential to interpret the results. Consistently across studies, sleep deprivation lasting longer than 24 h has been shown to inhibit cell proliferation in the hippocampus, independent of the methodology used or the sleep parameter targeted. Sleep deprivation of less than 24 h may actually increase cell proliferation. Effects on cell survival and maturation have been more variable across studies.
6.1 Short-Term Sleep Deprivation
Short-term sleep deprivation is defined here as procedures that last <2 days, as these appear to affect neurogenesis differently than do longer procedures. Three studies have investigated the effect of 12 h total sleep deprivation during the light phase on cell proliferation in the dorsal dentate gyrus, using BrdU, Ki67, or PCNA as proliferation markers. Two of the studies used rats and reported a significant increase (24 and 38 %, respectively) in cell proliferation using 2 h BrdU labeling (Grassi et al. 2006; Junek et al. 2010). Interpretation is complicated by the observation in one study that despite increased BrdU labeling, there was no change in labeling for the endogenous proliferation markers Ki67 or PCNA (Junek et al. 2010). This was taken to suggest that 12 h total sleep deprivation might transiently accelerate the cell cycle, allowing more dividing cells to enter S-phase (and incorporate BrdU) without increasing the total number of dividing cells (Junek et al. 2010). This is an intriguing hypothesis that merits further study using additional markers of cell cycle phase to measure cycle duration more directly. A study of 12 h total sleep deprivation in mice did not find a change in either 2 h BrdU or Ki67 (van der Borght et al. 2006). As the deprivation and labeling procedures matched those used in the two rat studies, a species difference is possible.
Four studies have examined the effect of 24 h sleep deprivation on BrdU or Ki67 labeling, using procedures for total sleep deprivation (Roman et al. 2005; Junek et al. 2010), REM sleep deprivation (Mirescu et al. 2006), or sleep fragmentation (Guzman-Marin et al. 2007a). Significant effects on cell proliferation or survival in the granule cell layer/subgranular zone were not observed, although one study did report a trend for reduced proliferation in the subgranular zone, and a significant reduction in the hilus (Roman et al. 2005). Also, the sleep fragmentation procedure reduced the number of new cells that expressed neuron-specific markers by ~20 %, indicating an effect on cell differentiation and maturation (Guzman-Marin et al. 2007a).
In addition to 12 and 24 h procedures, Junek et al. (2010) also examined 36 and 48 h total sleep deprivation. The transient increase in BrdU labeling observed following 12 h total sleep deprivation was absent following the other durations, and a trend for reduced cell proliferation was apparent in the 48 h total sleep deprivation group (Junek et al. 2010). One other study did report a significant reduction of cell proliferation in rats after 48 h of total sleep deprivation by gentle handling (Garcia-Garcia et al. 2011). Inhibitory effects of total sleep deprivation on hippocampal cell proliferation thus appear to emerge at about 2 days of sleep loss.
6.2 Long-Term Sleep Deprivation
The first long-term total sleep deprivation studies were conducted using rats and forced locomotion to prevent sleep for 56 h (state-contingent disc-over-water rotation) (Tung et al. 2005) or 96 h (intermittent treadmill) (Guzman-Marin et al. 2003). By contrast with locomotor control groups, the total sleep deprivation groups showed a reduction of 36 % (Tung et al. 2005) or 54 % (Guzman-Marin et al. 2003) in the basal rate of cell proliferation in the dentate gyrus, assessed by 2 or 48 h BrdU labeling, respectively. EEG recordings confirmed a >90 % reduction in total sleep time, with no REM sleep. Similar results have been obtained in mice using 96 h forced treadmill locomotion and 2 h BrdU labeling, but with a possible strain difference. Proliferation was reduced by 50 % in C57BL6 mice but by <15 % in C3H mice (n.s.) (Sompol et al. 2011). Cell proliferation was rescued in C57 mice but was not affected in C3H mice by administration of N-acetyl-serotonin (NAS), a precursor to melatonin that C57 mice lack. This was taken to suggest that endogenous NAS (or melatonin) might protect the neurogenic niche in the C3H strain from inhibitory effects of total sleep deprivation. However, rat strains showing a strong suppression of proliferation by total sleep deprivation are not deficient in NAS or melatonin.
Total sleep deprivation may affect neurogenesis beyond cell proliferation. A study using the 96 h treadmill procedure with 3-week survival after BrdU administration confirmed a 40 % reduction in BrdU labeling, and further reported a 47 % reduction in the proportion of new cells co-labeled with the neuronal marker NeuN (Guzman-Marin et al. 2005). This resulted in a net 60 % reduction of hippocampal neurogenesis, reflecting inhibition of cell proliferation, and a change in cell differentiation, survival, or both.
6.3 Sleep Restriction
Total deprivation of sleep beyond 1 day is presumably rare under natural conditions. By contrast, periods of sleep restriction of varying duration may be quite common, e.g., in shift work, or insomnia. To model chronic sleep restriction, rats were confined to a slowly rotating drum for 20 h/day, for 8 days, with 4 h recovery sleep permitted each day at light onset (Roman et al. 2005). BrdU was administered 5 days prior to sleep restriction. This procedure significantly reduced Ki67 labeling in the hilus and subgranular zone, but the effect in the subgranular zone was not significant by contrast with a forced activity control group, suggesting a nonspecific stress effect. No significant effects were observed in measures of cell survival or maturation, assessed by BrdU and NeuN co-labeling. Similar results were obtained in 30–61 day old adolescent rats subjected to the same 20 h/day sleep restriction procedure for 30 days, using BrdU labeling for cell survival and DCX for maturation (Novati et al. 2011). This study did not include a measure of cell proliferation.
One other study examined the effect of a 6 h/day sleep restriction (gentle handling and exposure to novel objects during the first 6 h of the light period) on neurogenesis stimulated in rats by training on a hippocampal-dependent spatial learning task (Hairston et al. 2005). Rats received two injections of BrdU 7 days prior to 4 days of water maze training. In control rats, training increased both cell survival (total BrdU) and the fraction of new cells expressing the neuronal marker DCX. In sleep-restricted rats, the training effect on cell survival and neurogenesis was absent, and spatial learning was impaired. These results imply that sleep following training is necessary for spatial learning, and that this may involve a sleep-dependent recruitment (and thus survival) of new neurons. The sleep restriction procedure was also associated with increased CORT , raising the possibility that the effects on neurogenesis and performance were mediated by a nonspecific stress response. The role of stress and CORT in effects of sleep manipulations is discussed further below.
6.4 Sleep Fragmentation
Sleep continuity and the sequences of stages are thought to be important for presumed restorative functions of sleep. Sleep fragmentation is associated with non-restorative sleep, and is characteristic of a variety of common conditions, including obstructive sleep apnea, depression, primary insomnia, and old age (Bonnet and Arand 2003). Two studies have examined the effect of experimental sleep fragmentation on hippocampal neurogenesis. One study confined rats to a treadmill that moved at 10 cm/s for 3 s once every 33 s for 1, 4, or 7 consecutive days (Guzman-Marin et al. 2007a). EEG recordings confirmed highly fragmented sleep characterized by shorter bout duration, increased bout number, a normal amount of NREM sleep, and a ~80 % decrease in REM sleep. Cell proliferation (2 h BrdU or Ki67) was unaffected by the 1 day procedure, but was reduced ~70 % in the 4 and 7 day conditions, compared to treadmill control rats. The proportion of new cells expressing a neuronal phenotype (3-week BrdU+NeuN) was significantly reduced in all 3 conditions. Notably, the 52 % reduction observed in the 4-day sleep fragmentation condition exceeded the 36 % reduction previously observed in rats subjected to 4-day total sleep deprivation (Guzman-Marin et al. 2003). The results leave open the possibility that these effects were mediated by suppression of REM sleep rather than sleep fragmentation.
A second sleep fragmentation study from the same group used a modified treadmill activation protocol to examine effects of sleep fragmentation on neurogenesis and subsequent hippocampal function (Sportiche et al. 2010). The treadmill was actuated for 3 s after 30 consecutive seconds of NREM sleep. This induced sleep fragmentation without altering total daily NREM or REM sleep. BrdU was administered on days 4 and 5 of the 12-day sleep fragmentation procedure, which was followed by 14 days of recovery and 7 days of training on a hippocampus-sensitive Barnes maze spatial learning task. The sleep fragmentation procedure decreased BrdU cell counts by 30 % and impaired performance, but did not decrease the proportion of BrdU-ir cells coexpressing NeuN relative to yoked and treadmill control groups. This latter result suggests that the decrease in the proportion of double-labeled cells observed previously in response to sleep fragmentation (Guzman-Marin et al. 2007a) was due to the suppression of REM sleep caused by the sleep fragmentation procedure. Alternatively, the 7-day training procedure that followed sleep fragmentation may have offered protection.
6.5 REM Sleep Deprivation
Total sleep deprivation and sleep fragmentation procedures leave open the question of whether sleep stages contribute differentially to the suppression of neurogenesis. Selective deprivation of NREM sleep is not possible without affecting REM sleep, because REM sleep is normally entered through NREM sleep. However, a relatively selective REM sleep deprivation can be achieved by state dependent or preemptive deprivation techniques. A classic preemptive REM sleep deprivation method exploits the muscle atonia that accompanies REM sleep. In this procedure, rats or mice are placed on a small platform in a pool of water. Loss of muscle tone at REM sleep onset causes the animal to dip its head toward the water, aborting the episode. This method almost completely suppresses REM sleep, and typically spares 60 % or more of NREM sleep, depending on the duration of the procedure. Multiple platforms can be used to permit social housing and locomotion. Two studies using the platform method reported a consistent 40–50 % reduction of cell proliferation following REM sleep deprivation of 72 h (Mirescu et al. 2006) or 96 h (Mueller et al. 2008). The latter study showed the effect to be independent of the REM sleep deprivation method (single vs. multiple platform), the housing condition (single vs. group housing), the proliferation marker (2 h BrdU vs. Ki67), and the rat strain (Sprague–Dawley vs. Long Evans), and to include both the granule cell layer/subgranular zone and hilus.
A more selective, albeit less complete REM sleep deprivation can be accomplished using a state-contingent deprivation procedure, in which an arousal stimulus is applied after REM sleep is detected. One study used EEG recordings with automated real-time sleep staging to identify REM sleep and actuate a treadmill (Guzman-Marin et al. 2008). Yoked control animals were stimulated at the same time, and allowed to sleep when the REM sleep deprivation animals were spontaneously awake. This method reduced REM sleep by ~80 % in the REM sleep deprivation group relative to yoked and home cage controls. NREM sleep was reduced by only 17 % and NREM EEG slow wave activity was not reduced, as would be expected if there was a homeostatic deficit in this sleep stage. Cell proliferation (BrdU and Ki67) in the dentate gyrus was reduced by 63 % in REM sleep deprivation rats compared to the yoked controls, and across all rats the number of new cells correlated +0.84 with the amount of REM sleep obtained, but only +0.26 with the amount of NREM sleep. These observations support an important role for REM sleep in sustaining basal levels of cell proliferation in the hippocampus.
By contrast with effects on cell proliferation, reported effects of REM sleep deprivation on maturation are less consistent across studies. The two studies that used the platform REM sleep deprivation method reported no effect of 72 or 96 h REM sleep deprivation on the proportion of new cells expressing the neuronal markers TuJ1, NeuN, or DCX (Mirescu et al. 2006; Mueller et al. 2008). However, the 96 h REM sleep deprivation study that used the more selective state-contingent REM sleep deprivation method observed a 30 % reduction in the proportion of new cells co-labeled with NeuN, relative to home cage controls (Guzman-Marin et al. 2008). The reduction was only 10 % relative to yoked controls, but this likely reflects the 30 % reduction in REM sleep sustained by the yoked controls relative to home cage controls. Although the discrepancy in reported effects on cell maturation remains to be clarified, all three REM sleep deprivation studies are consistent in observing a reduction in cell proliferation comparable to that observed following total sleep deprivation procedures.
6.6 Regional Differences
An important question is whether sleep deprivation affects adult neurogenesis equivalently throughout the brain, or whether the effects are regionally specific. Regional differences could provide clues to the mechanisms by which sleep loss affects neurogenesis, as well as internal controls for nonspecific effects of sleep deprivation procedures that may affect neurogenesis. There appears to be a consensus that cell proliferation in the subventricular zone is not affected by either short-term or long-term sleep deprivation procedures (Grassi et al. 2006; Mirescu et al. 2006; Junek et al. 2010). Therefore, effects of sleep deprivation on proliferation appear to be specific to the hippocampus, similar to other behavioral manipulations known to regulate hippocampal neurogenesis (e.g., training on hippocampal-dependent learning tasks).
Related posts:
 Memory Consolidation in Healthy Aging and Mild Cognitive Impairment
Restricted or Disrupted Sleep as a Causal Factor in the Development of Depression
Memory Consolidation in Healthy Aging and Mild Cognitive Impairment
Restricted or Disrupted Sleep as a Causal Factor in the Development of Depression
 and Electrophysiological Correlates of Sleep and Sleep Homeostasis
and Electrophysiological Correlates of Sleep and Sleep Homeostasis
 Treatment of Sleep Disorders and Its Relationship with Neuroplasticity
Treatment of Sleep Disorders and Its Relationship with Neuroplasticity
 and Synaptic Homeostasis
and Synaptic Homeostasis
 Studies of Sleep and Memory in Humans
Studies of Sleep and Memory in Humans
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


