A Simplified Regulatory Model
In characterizing the structure of the weight regulatory system, investigators classically have identified two regulatory components: a short-term component that controls the onset and cessation of feeding on a meal basis, and a long-term component that monitors and regulates body nutrient stores over extended periods (8–10). In theory, these two components modulate intake such that body weight is maintained, with minimal fluctuations, within a stable range. Thus, although having distinct mechanisms, these components act in an integrated fashion within the central nervous system (CNS) in performing regulatory corrections of body weight. Specifically, the potency of the short-term signals controlling meal-taking is decreased or enhanced within the CNS in response to long-term regulatory influences reflecting the state of nutrient stores, and feedback signals from the gut generated by the arrival and processing of nutrients.
Short- and Long-Term Feedback Signals
As represented in Figure 8-1, feeding is stimulated by both appetitive stimuli in the environment (sight, smell, taste of food), and internal biological stimuli arising from the regulatory system itself. Feeding provides energy substrates to the body, which first interact with the oral cavity and the gastrointestinal tract to provide sensory (taste, distension) and humoral (peptide) feedback signals, which are correlates of nutrient appearance and amount. A majority of these signals are transmitted to afferent centers in the hindbrain, where they are processed, providing the basis for both input to higher-level integrating centers in the hypothalamus, and efferent signals controlling feeding and the activity of the gastrointestinal tract. This is the short-term component of the regulatory system. Feedback to the hypothalamus from the short-term component is in turn integrated with stimulation from long-term regulatory signals, providing the basis for a homeostatic response.
A large proportion of ingested calories are used by the body for maintenance and activity. The unused portion of ingested substrate is processed and stored primarily as depot fat. The size of body fat mass, or energy stores, is represented by hormonal, nutrient, and as newer evidence indicates, sensory feedback signals, which comprise the long-term component of the system. These signals ideally are reliable correlates of the amount of stored body fat, and serve as error signals to a network of hormone and energy-sensing neurons in the hypothalamus. Energy homeostatic influences emanating from this network are transmitted via descending pathways to the hindbrain, where, as noted above, they provide excitatory or inhibitory influences on feeding and gastrointestinal mechanisms controlled there.
Integration of Long- and Short-Term Signals
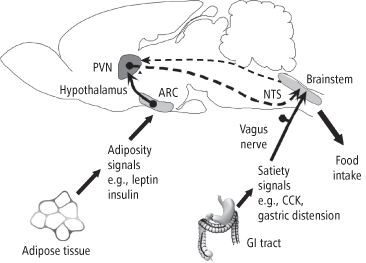
A representation of the integrated action of the short- and long-term components can be seen in Figure 8-2, which shows two hypothalamic areas involved in processing long-term feedback signals, the arcuate nucleus (ARC) and the paraventricular nucleus (PVN), as well as one hindbrain center, the nucleus of the solitary tract (NTS), which receives incoming stimulation from the gastrointestinal tract. Stimulation arriving at the NTS is sent via ascending fibers to the hypothalamus, where it is integrated with long-term regulatory signals acting there. Descending fibers project regulatory signals from the hypothalamus to the NTS, which, together with other hindbrain structures, coordinate signals controlling meal-taking behavior (11). An excellent example of this integrative pathway is the ability of insulin, a long-term regulatory signal, to enhance the feeding-inhibitory potency of cholecystokinin (CCK), a short-term satiety signal (12). Before we discuss specific long- and short-term signals and their processing in the next sections, it should be noted that there are some exceptions to the model presented above. For example, two peptides of gastrointestinal origin, ghrelin and peptide YY3-36 (PYY(3-36)), appear to stimulate hypothalamic centers directly (13). Leptin, a long-term regulatory signal whose primary action is on hypothalamic centers, also binds to areas of the hindbrain and influences satiety signaling there (14–15). Nevertheless, our model will serve as a useful tool for the analysis of the many signals arriving at the CNS, and their multiple modes of action.
Figure 8-2 Integration of long- and short-term feeding signals. Afferent stimulation arriving at the NTS from the gastrointestinal tract and oral cavity is sent via ascending fibers to the hypothalamus, where it is integrated with long-term regulatory signals from peripheral sources acting there. Descending fibers project regulatory influences from the hypothalamus to the NTS, which, together with other hindbrain structures, coordinate efferent signals controlling meal-taking behavior.
(Adapted and reprinted from Morton GJ, Blevins JE, Williams DL, et al. Leptin action in the forebrain regulates the hindbrain response to satiety signals. J Clin Invest 2005; 115: 703–710, with permission)
Long-Term Weight Regulatory Signals
Leptin and Insulin
The adipose tissue-derived hormone leptin and the pancreatic hormone insulin both serve as long-term feedback signals to the CNS representing the status of body fat stores. The existence of a humoral factor released by adipocytes and representing body fat stores was first proposed in 1953 (16) and was supported by studies showing that obesity resulting from an autosomal recessive mutation in ob/ob mice could be reversed when their circulation was crossed with that of wild-type lean littermates (17–18). The actual discovery of the hormone leptin in 1994 using ob/ob mice awaited the development of sophisticated molecular biological techniques (19). A role for insulin as a feedback signal representing body fat was first hypothesized in 1974 (20), and convincing behavioral and biological evidence has since been generated for this function of the hormone (21–22). It should be noted that although insulin’s actions in the periphery are anabolic, its effects in the brain are catabolic and similar to that of leptin, to inhibit feeding and reduce body weight.
Both leptin and insulin meet the criteria for an adiposity feedback signal (23). Both hormones circulate at levels proportional to body fat mass (24–25), and cross the blood–brain barrier via active transport mechanisms (26–27). Receptors for both hormones are found in hypothalamic areas involved in energy homeostasis, most notably the arcuate nucleus of the hypothalamus (28–29). The administration of either hormone directly into the brain reduces food intake (30–32), while inhibition of their effects in the brain stimulates feeding (33–35). In addition, both leptin and insulin stimulate energy expenditure via the melanocortin system (described below), which when activated may have weight-reducing effects independent of feeding inhibition (36–38). It should be noted that leptin is also expressed in the gastric mucosa (39) and appears to play a role in stimulating satiety by modulating incoming vagal stimulation in response to the arrival of nutrients in the stomach (40–41).
Hormone Signal Processing in the Hypothalamus
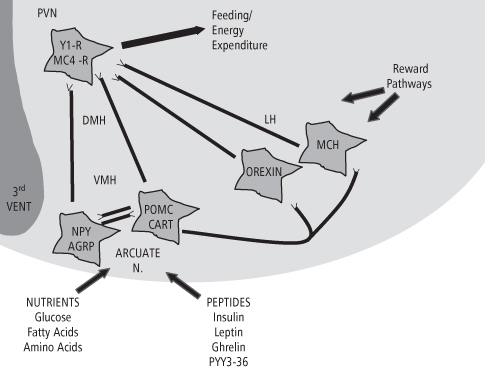
The identification of leptin and insulin as feedback signals for energy homeostasis has greatly expanded our understanding of the hypothalamic system that participates in the regulation of feeding and body weight. Figure 8-3 represents the ventral aspect of the hypothalamus adjacent to the third ventricle and shows three major centers involved in homeostatic signal processing: the ARC, PVN, and lateral hypothalamic area (LHA). Receptors for leptin and insulin are found on two distinct sets of neurons in the ARC, identified as NPY/AgRP (Neuropeptide Y/Agouti gene-Related Peptide) and POMC/CART (Pro-opiomelanocortin/Cocaine- and Amphetamine-Related Transcript), which reciprocally inhibit each other and also send projections to the PVN. These two sets of neurons are considered first-order neurons, since they respond directly to incoming hormonal signals. Stimulation of leptin and insulin receptors inhibits the expression of NPY and AgRP, two orexigenic (feeding-stimulatory) neuropeptides found in these first-order neurons, and stimulates the expression of POMC and CART, two anorexigenic (feeding-inhibitory) peptides also found in these neurons (23). NPY plays a major role in feeding stimulation (42), and chronic microinjection of NPY into the PVN rapidly induces hyperphagia and obesity in normal rats (43). Centrally-administered AgRP and its agonists also potently increase food intake in rats (44–45). Conversely, centrally administered CART and α-melanocyte-stimulating hormone (α-MSH, the active cleavage product of POMC) potently reduce feeding and body weight in rodent models on both a short- and long-term basis (46–48). Both AgRP and α-MSH are components of the hypothalamic melanocortin system (reviewed below). Both sets of ARC neurons project to the PVN and other second-order hypothalamic and brainstem centers, in this case signaling inhibition of feeding. When hypothalamic leptin and insulin stimulation is reduced, the reverse effect on neuropeptide expression in these ARC neuron sets takes place, resulting in feeding stimulation. Thus, leptin and insulin drive a homeostatic system that sets the “tone” for feeding stimulatory and inhibitory influences.
Figure 8-3 First order and higher order hypothalamic systems participating in the regulation of feeding and body weight. Peptide and nutrient signals act on first order NPY/AGRP and POMC/CART neurons in the arcuate nucleus of the hypothalamus, which have reciprocal axon projections to each other. These sets of neurons project to the paraventricular nucleus (PVN), which contains NPY and melanocortin receptors involved in signaling feeding and energy expenditure. NPY and POMC neurons also project to orexin and MCH neurons in the lateral hypothalamus (LH), and modulate their feeding-stimulatory influences on the PVN. Reward-mediating tracts synapse with neurons in the LH, providing feedback which is integrated into the stimulatory input to the PVN. The dorsomedial (DMH) and ventromedial (VMH) hypothalamic areas contain receptors for many feeding-related neuropeptides, and participate in the processing of incoming and outgoing homeostatic signals.
(Adapted and reprinted with permission from Schwartz, MW, Morton, GJ. Obesity: keeping hunger at bay. Nature 2002; 418: 595–597)
Higher-Order CNS Systems Involved in Energy Homeostasis
The hypothalamus not only receives and integrates incoming hormonal signals representing peripheral energy stores, but sends impulses to and receives input from higher-order integrating systems in the CNS that participate in energy homeostasis. In some cases, these systems act as second-level integrators and effectors for feeding and metabolic signals originating in first-order neurons, such as the melanocortin system within the hypothalamus. In other cases, higher-order systems themselves provide feeding stimulatory or inhibitory influences that impinge on ongoing hypothalamic signaling, such as lateral hypothalamic orexigenic neuropeptides and the dopamine and opioid reward systems. We shall review the major higher-order feeding-related CNS systems next.
The Hypothalamic Melanocortin System
Independently of work on the roles of leptin and insulin, other investigators in the early 1990s were pursuing the basis for obesity in the agouti mouse, which bears an autosomal-dominant mutation resulting in yellow coat color, adult onset obesity, and enhanced linear growth. The yellow coat color (seen only in the mutants) was shown to be the result of the ectopic expression of a mutated form of melanocortin protein, termed agouti protein, acting as an antagonist at the melanocortin 1 receptor (MC1-R), which controls pigmentation (49–50). Binding of the agouti protein as an antagonist to two melanocortin receptors later discovered in the brain, MC3-R and MC4-R, was shown to be the cause of the obesity in agouti mice (51–52). The feeding-stimulatory effect of the related peptide AgRP, whose expression in the hypothalamus is inhibited by leptin and insulin in NPY-AgRP arcuate neurons, is a result of its ability to act as an antagonist at MC4-R receptors in the PVN (53).
Further work identified the agonist for these receptors, capable of potently inhibiting feeding, as α-MSH, the active cleavage product of POMC expressed in hypothalamic POMC-CART neurons when stimulated by leptin and insulin (54). Projections from these neurons carry α-MSH to the PVN, where MC4-R receptors are expressed, and also travel to sympathetic outflow neurons in the spinal cord, mediating peripheral lipolysis and energy expenditure (55–56). Thus the melanocortin system serves as a mediator of the effects of leptin and insulin on ARC neurons, with the summation of its peptide messengers α-MSH and AgRP determining the effect of the system on the PVN-feeding and sympathetic outflow pathways, ultimately controlling both energy intake and energy expenditure. Of interest is the fact that the melanocortin system represents a virtually unique case in neuronal physiology in which both endogenous agonists and antagonists are expressed and act on the same receptor (57). Mutations of the melanocortin system resulting in human obesity are less rare than those of the leptin signaling system, accounting for an estimated 4-5% of severe human obesity (58).
Lateral Hypothalamic Orexigenic Neuropeptides
The Orexins/Hypocretins
While the roles of two orexigenic neuropeptides within first-order arcuate neurons were under intense investigation (NPY and AgRP), two additional feeding-stimulatory neuropeptides expressed in the LHA were discovered, which broadened our understanding of homeostatic mechanisms within the hypothalamus (see Figure 8-3). The orexins (orexin-A and orexin-B, also called hypocretin-1 and hypocretin-2), along with their receptors, were discovered in the LHA of the rat brain in 1998 (59–60). When centrally administered, orexin-A appears to be the more potent of the two forms for feeding stimulation, (61), but its feeding-stimulatory effects are less profound than those of central NPY (62). Orexin-A and its receptor are highly expressed in the hypothalamus in response to caloric deprivation (63). Nevertheless chronic central administration of orexin-A elevates light-cycle food intake only, with no change in 24-hour calorie intake or body weight. This observation has led to the notion that the neuropeptide plays a role in prolonging feeding or delaying meal-related satiety (64–65). Further work on the function of the orexins indicates that the peptides play a more complex role in energy homeostasis than previously thought, involving interactions between nutritional status and sleep, arousal, and possibly reward (66–68). However, extensive reciprocal connections between orexin and arcuate NPY and POMC neurons (69–70) and the presence of leptin receptors on orexin-expressing neurons in the LHA (71) indicate that the neuropeptide is also involved in at least the short-term control of feeding behavior.
Melanin-Concentrating Hormone
A role for the hypothalamic peptide melanin-concentrating hormone (MCH) in feeding behavior was first identified in 1996 in a study showing increased feeding and decreased energy expenditure in rats and mice following central microinjection of the peptide (72). Similar to orexin-A, MCH expression in the LHA increases with fasting (72), but MCH appears to play a more prominent role in the control of energy homeostasis. Central administration to rodents induces hyperphagia in both the light and dark phases, and MCH overexpression in transgenic mice leads to obesity and insulin resistance (73). Conversely, MCH knockout mice are hypophagic and have reduced body weight and body fat due to increased metabolic rate (74). MCH receptors are found in several brain regions (75), but targeted deletion of the MCH-1R receptor in the LHA also results in lean mice due to increased energy expenditure, despite the fact that the MCH receptor-null mice paradoxically become hyperphagic (76). Like the orexins, the MCH system appears to play a more complex role in appetitive behavior, with MCH neurons making widespread connections with several brain areas involved in taste, olfaction, and visceral sensation (77–78). Also like orexin neurons, cells expressing MCH receive input from POMC-CART and NPY-ARC neurons (79), and when stimulated by orexin from closely associated LHA sites, MCH neurons are activated (80).
Hypothalamic Systems Involved in Food Reward
One significant aspect of feeding behavior is that it appears to be driven not only by the onset of hunger and the availability of food items, but also by the rewarding, or hedonic effects, of food consumption. The CNS houses at least three separate systems that mediate food reward and interact with energy homeostatic pathways that control feeding. Indeed, it is in part through these systems that palatable high-energy diets exert their well-known obesogenic effects when offered ad libitum to rodents. Reward-based systems in the brain that influence regulatory feeding circuits have been termed non-homeostatic, to distinguish them from energy-based homeostatic feeding systems in the hypothalamus and hindbrain (81). This is a crucial concept, since it implies that there are CNS processes that can alter or override homeostatic feeding controls. Anatomically, feeding reward-related pathways have been shown to project to the lateral hypothalamic area (82), among other sites, where they can interact with energy-based feeding mechanisms. We shall consider these reward-based systems next.
The Dopamine System
The dopamine system is widespread in the midbrain and forebrain and participates in the control of numerous behaviors such as feeding, arousal and motor function (83). It is well established that dopamine mediates both the motivating and rewarding aspects of food consumption, although dopamine’s rewarding vs. motivational effects in the maintenance of feeding are still being delineated (84). For example, under dopamine antagonist blockade food loses its normal ability to maintain responding in well-trained animals (85), and the animals respond to given concentrations of saccharin and sugar solutions as if they were weaker (86–87). Because, under these conditions, consumption of more palatable food items would be required to achieve an adequate level of food reward, decreased dopamine signaling in the hypothalamus of obese animals and humans has been suggested as a potential cause of obesity (88–89). Consistent with this hypothesis is the recent observation that administration of a drug that enhances brain dopamine levels decreases energy intake, and fat intake specifically, in normal-weight humans (90). Also consistent with this notion is the fact that food restriction reduces dopamine levels in the nucleus accumbens (91), while food reward elevates dopamine levels in this forebrain structure (92). These latter effects are proposed to underlie a new model of food addiction based on intermittent sugar consumption in rats (93). Indeed, recent neuroimaging studies have identified common neuronal circuits modulated by dopamine, which may be shared in states of addiction and obesity (94). Overall, the effects of manipulations of hypothalamic and forebrain dopamine levels on motivation and reward are complex, although the data suggest that dopamine plays a significant role in stimulating overeating and maintaining obesity (95).
The Opioid System
The opioid system has long been known to influence feeding behavior (96), although a clear understanding of its role, and of the neural substrates that mediate it, has developed only gradually over the years (97). Three endogenous opioid peptides—β-endorphin, encephalin, and dynorphin—differentially interacting with three receptor classes (98) are involved in opioid control of feeding (97). Three major hypotheses have been advanced for how the opioids influence feeding, with experimental evidence supporting all three. Opioids appear to participate in the process of orosensory food reward, in the control of macronutrient consumption, and in determining meal duration (99). Thus, administration of opioid agonists preferentially enhances intake of palatable non-nutrient or nutrient solids and liquids (100–101), specifically increases fat intake (102–103); and, as demonstrated with opioid antagonists, appears to prolong meals (104–105). It is likely, however, that these proposed functions interact with pre-existing preferences and nutrient availability in determining opioid effects on feeding (106–107). With regard to brain regions that mediate opioid-feeding effects, the opioids appear to act on feeding-related pathways at multiple CNS loci, including the NTS, PVN, nucleus accumbens, and central nucleus of the amygdala. Opioid receptors have been found at all four sites, and specific opioid effects can be demonstrated at each locus (99). Studies are underway to determine exactly how neurons of this distributed neural system interact to generate opioid feeding effects. That this system plays a prominent role in at least the maintenance of obesity is demonstrated by the effects of a potent opioid receptor antagonist in diet-induced obesity (DIO) rats, including reduced body weight and body fat, and enhanced lipid oxidation (108).
The Endocannaboid System
The endocannabinoid system is a relatively recent addition to the list of reward systems influencing feeding. Although the appetite-inducing effects of cannabis have long been known, the first cannabinoid receptor (CB-1) was not identified until 1990 (109), and ligands for this and a second receptor (CB-2) were not identified until the mid-1990s (110–111). The two primary endogenous ligands for CB receptors are the fatty acid derivatives anandamide and 2-arachidonoyl glycerol (2-AG). Administration of these receptor ligands stimulates feeding even in sated animals (112–113), while pharmacological blockade of the CB-1 receptor results in inhibition of intake of both standard chow and more palatable diets (114–115). Moreover, knockout of the CB-1 receptor in mice leads to hypophagia following fasting, decreased fat mass, resistance to diet-induced obesity, and enhanced leptin sensitivity (116–117). Finally, levels of the endocannabinoids decrease during feeding and increase during fasting (118). The CB-1 receptor, found in both the brain and periphery, is responsible for all of these effects. Two hypotheses have been offered to explain the effects of endocannabinoids on food intake: endocannabinoids may act first by increasing the motivation to find and consume highly palatable food items (by enhancing food reward), and second, by modulating hypothalamic levels of orexigenic and anorexigenic neuropeptides (119). These hypotheses are supported by the widespread presence of endocannabinoid and CB-1 receptors in central mesolimbic pathways involved in reward (ventral tegmental area and nucleus accumbens) (120) and in medial and lateral hypothalamic neuron subpopulations expressing orexin-1, MCH, and CART (121). In addition, leptin levels have themselves been found to regulate the expression of hypothalamic endocannabinoids (116), linking endocannabinoid action to peripheral and central homeostatic signaling.
Additional Hypothalamic Influences
We shall consider several additional higher-order neuropeptides that play important roles in influencing feeding signaling in the hypothalamus. Although the precise manner in which these factors are integrated into known energy homeostatic pathways has not been defined, they are of major interest since their expression in the brain is determined at least in part by levels of the two major regulatory hormones, leptin and insulin. Moreover, with the exception of galanin and norepinephrine, the feeding effects of these neuropeptides appear to be mediated by the leptin–melanocortin system.
Galanin and Norepinephrine
Galanin is a 29 amino acid peptide widely distributed throughout the gastrointestinal tract and the brain. High concentrations of galanin and its receptors are found in the hypothalamus (122), and microinjection of nmole amounts of this neuropeptide into the PVN is found to potently stimulate food intake in non-deprived rats (123). The effects of microinjected galanin on feeding are brief, however (less than 30 minutes), suggesting that its role may be limited to meal-initiation (124). Thus, chronic central administration of galanin for 14 days increased food intake primarily during the light cycle, but did not lead to increased body weight (125). Although initial observations indicated that galanin selectively stimulated fat intake (126), subsequent studies have shown no specific macronutrient preference following galanin administration (127–128).
Galanin may exert its feeding effects by interacting with norepinephrine (NE), another hypothalamic neuropeptide that stimulates feeding when administered to the PVN (129). Indeed, chronic microinjection of NE into the PVN results in hyperphagia and elevated body weight in rats (130). Both galanin and NE are expressed in midbrain neurons that project to the PVN (131–132), and both target the parvicellular region of the PVN to exert their feeding-stimulatory effects (133). Galanin appears to stimulate feeding via α2-noradrenergic receptors, since injection of α2-receptor antagonists into the PVN abolishes galanin-induced feeding (134). Although galanin’s feeding effects appear to be limited to meal initiation, central administration of both leptin (135) and insulin (136) reduces hypothalamic galanin expression, indicating that galanin is a target of hormone signaling in the hypothalamus and may participate in the central control of appetite (137).
Serotonin and Its Receptors
Serotonin (5-hydroxytryptamine, 5-HT) has long been known as a feeding-inhibitory neurotransmitter acting at multiple hypothalamic sites, including the PVN, VMH, and DMH (138). The serotonin system is widespread in the brain, however, and at least 14 distinct 5-HT receptors have been identified (139). Notably, the serotonin system is also implicated in reward processes, but primarily in relation to mediating the effects of psychoactive drugs (140–141). There is strong evidence that at least three 5-HT receptors, 5-HT1A, 5-HT1B, and 5-HT2C mediate serotonin’s effects on feeding (142), with convincing evidence favoring the 5-HT2C receptor as the primary receptor involved (140). Thus, 5-HT2C receptor mRNA is highly expressed in POMC neurons of the ARC nucleus (143), and serotonin agonists cause the release of α-MSH from hypothalamic slices (144). Moreover, 5-HT2C knockout mice are hyperphagic, obese, and respond poorly to the feeding-inhibitory properties of d-fenfluramine, a serotonin agonist (145–146). The specific behavioral effect of serotonin appears to be stimulation of the satiety process, since administration of serotonin agonists to rats reduces meal size and meal duration, but not meal onset or meal number (147–148). Moreover, there is evidence that serotonin reduces appetite specifically for dietary carbohydrate (149–150). Finally, the potency of 5-HT2C receptor activation for feeding inhibition has led to the development of numerous anti-obesity drugs that act as 5-HT2C agonists (151–152), or act by prolonging serotonin availability in the hypothalamus by inhibiting its reuptake (153).
Corticotropin-Releasing Factor and Urocortin
Corticotropin-releasing factor (CRF), a neuropeptide that stimulates the release of adrenocorticotrophic hormone (ACTH) and POMC from the pituitary, was initially known for its role in mediating the physiological response to stress (154). However, CRF was later found to inhibit feeding behavior when centrally injected (155) and the PVN was identified as the major site of the anorectic effects of this peptide (156). More recently, another neuropeptide in the CRF family, urocortin, was identified (157), and was found to be a much more potent inhibitor of feeding than CRF itself, in the absence of behavioral markers of stress (158). Urocortin’s feeding-inhibitory effects have been localized to the ventromedial hypothalamic nucleus (VMH) (159). Thus, CRF and urocortin appear to inhibit feeding via hypothalamic mechanisms and may exert their effect specifically on NPY-responsive neurons (160). Although CRF and urocortin bind to both CRF-1 and CRF-2 receptors, the latter receptor is believed to be the primary mediator of the anorexic effects of these peptides, since urocortin binds with much higher affinity to the CRF-2 receptor (158). Consistent with its feeding-inhibitory effects, expression of CRF and its receptors is reduced in the hypothalamus of food-deprived and obese rats (161–162), and elevations of leptin increase the expression and release of CRF in the PVN (163–164). Thus, CRF and urocortin may participate in leptin’s feeding-inhibitory effects in the hypothalamus (165–166).
Short-Term Feeding Control Signals
In addition to the complex CNS energy homeostatic systems described above, the gastrointestinal (GI) tract and associated organs, liver and pancreas, give rise to numerous peptide and/or afferent neural signals acting at the hindbrain and/or hypothalamus, which signal short-term nutrient status and participate in the control of feeding. As noted earlier in this chapter, the NTS in the hindbrain integrates descending hypothalamic and incoming peripheral sensory information to control meal size. Additional hindbrain areas involved in the control of feeding include the area postrema (AP) and the dorsal motor nucleus of the vagus nerve (DMX); these structures, together with the NTS, are collectively known as the dorsal vagal complex (167).
The dorsal vagal complex (Figure 8-4) receives afferent stimulation from the periphery via the 10th cranial nerve, or vagus nerve, which terminates in the NTS. Other cranial nerves relay taste and mechanical stimulation from the oral cavity. The dorsal vagal complex can also be stimulated directly by hormones and metabolites, since the AP lies adjacent to the 4th ventricle of the brain, and contains a porous blood–brain barrier (168). The AP and NTS reciprocally innervate each other and project to forebrain regions involved in the control of feeding. Efferent impulses to the GI tract and associated organs are relayed to the periphery via the DMX. While most of the incoming peripheral signals reflect short-term nutrient status, their effect on feeding may be short or long-lasting. We will begin our review with peptides of gastrointestinal origin, represented, along with pancreatic factors, in Figure 8-5.
Figure 8-4 Schematic cross-section of the Dorsal Vagal Complex at the level of the area postrema (AP) in the hindbrain. Sensory input from the gastrointestinal tract and oral cavity arrives at the NTS and is relayed to the hypothalamus, which in turn integrates this information and provides descending influences which alter the activity of the dorsal motor nucleus of the vagus nerve (DMX). The DMX and other motor centers control the outflow of efferent impulses signaling feeding, energy expenditure and gut motility to peripheral tissues and organs. Direct hormonal stimulation of receptors in the walls of the 4th ventricle by circulating peptides involved in both the long- and short-term control of feeding also influences the activity of the centers in the Dorsal Vagal Complex. CC, central canal.
(Adapted and reproduced with permission from Luckman SM, Lawrence CB. Anorectic brainstem peptides: more pieces to the puzzle. Trends Endocrinology Metab 2003;14: 60–5.). (ref. 167)
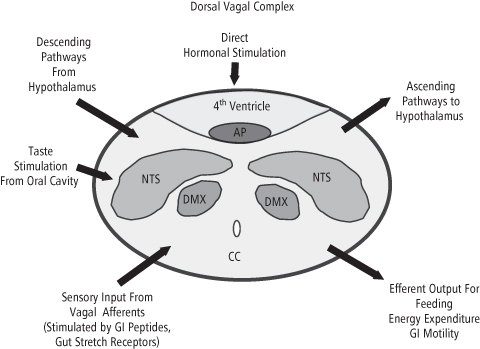
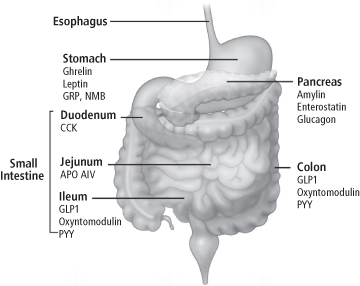
Figure 8-5 Sites of expression and release of gastrointestinal and pancreatic peptides involved in the regulation of food intake. Many, but not all of these peptides are involved in the short-term control of meal-taking. Leptin is included here since it is expressed in the mucosa of the stomach, and may play a role in modulating afferent feedback from the gut to the NTS. In addition to insulin, GLP-1, PYY3-36 and Apo A-IV may have longer-term influences on feeding regulation.
(Reproduced with permission from Cummings DE, Overduin J. Gastrointestinal regulation of food intake. J Clin Invest 2007; 117: 13–23.)
Gastrointestinal Peptides
Cholecystokinin
Of the peripheral satiety-related peptides, cholecystokinin (CCK) was the first to be discovered (169) and has been the most intensively studied. Although there are many biologically active forms of CCK (170), CCK-8, found primarily in the brain, and CCK-33, found in the circulation, are of most interest since they dose-dependently stimulate meal-related satiety. Thus, peripheral administration of CCK produces reductions of meal size in rodents and in lean and obese humans (171–173). The short-term nature of CCK’s action is demonstrated by the finding that chronic administration of CCK reliably decreases meal size, but results in no overall decrease in total food intake (174). However, the importance of this peptide in energy balance is seen in the finding that rats lacking the peripheral CCK receptor show increased meal size and gradually become obese (175). CCK is secreted into the enteric circulation from duodenal cells when food appears in the lumen. Dietary fat and protein potently stimulate CCK release, while carbohydrate does not (176). Peripheral CCK receptors (CCK1-R) are expressed on vagal afferent fibers innervating the exterior surface of the stomach and proximal intestine, and afferent signaling by CCK is mediated by the vagus nerve (177–178). Moreover, vagal afferent fibers responsive to gastric distention respond at a greater rate when CCK is administered, suggesting a synergistic effect of these two gut afferent signals (179–180). However, CCK1-R receptors are also found in the hindbrain, and lesions of the AP and NTS attenuate CCK-induced feeding inhibition (181), suggesting that CCK may also act directly on the hindbrain to induce satiety. Further, CCK microinjected into the hypothalamus also reduces feeding (182), presumably via CCK2-R brain receptors, since CCK2-R knockout mice are hyperphagic and become obese (183). A variety of CCK analogs have been developed for human weight loss, although human trials with CCK have not met with the same success as those conducted in animals (184).
Glucagon-Like Peptide-1 (GLP-1), PYY3-36 and Oxyntomodulin
Both GLP-1 and PYY3-36 are gut peptides released into the circulation from the L-cells of the distal intestine following a meal, with levels remaining elevated for relatively long periods following release (185–186). Both of these peptides, and the GLP-1 receptor agonist exendin-4, have been shown to inhibit feeding following peripheral administration in animals and humans (187–190). The site of action of GLP-1 and PYY3-36 for satiety effects remains under investigation. PYY3-36 is believed to act directly on hypothalamic Y2 receptors by inhibiting NPY neurons in the ARC (191), and this view is supported by the fact that PYY3-36 does not inhibit feeding in Y2 knockout mice (187). Similarly, central administration of GLP-1 potently reduces feeding, and GLP-1 responsive neurons are found not only in the ARC but in the NTS, suggesting that GLP-1 may bind to receptors in both of these areas (192–193). Moreover, the two peptides appear to work additively at the hypothalamic level in reducing food intake (194). Further work, however, has shown that surgical vagatomy can attenuate the feeding inhibitory effects of both GLP-1 and PYY3-36 (195), indicating that these peptides normally signal via peripheral receptors, with the information relayed by the vagus nerve. Thus, the precise mechanism underlying the satiety action of these peptides remains uncertain (196). Finally, GLP-1 and PYY3-36 exert their feeding-inhibitory effects via reductions of average meal size and increases of inter-meal intervals, rather than through immediate effects on meal termination (197–198). This longer-term aspect of the peptides’ action may hold therapeutic promise. Thus, chronic treatment with PYY3-36 has been shown to reduce food intake and adiposity in rodents (199–200), and human obese subjects remain responsive to the peptide’s feeding-inhibitory effects (201). Chronic administration of a long-acting form of GLP-1 reduces body weight in both lean and obese rats (198), and exendin-4, the GLP-1 agonist, has been found to cause significant weight loss in obese diabetic patients (202).
Oxyntomodulin (OXM) is derived from the same proglucagon precursor as GLP-1 and, like GLP-1, is secreted from intestinal L-cells in response to ingested calories (203). When administered acutely, OXM inhibits feeding acutely in both rats and humans (204–205), while chronic administration to rats reduces calorie intake and body weight gain (206). However, GLP-1 and OXM have dissimilar effects on feeding and body weight with regard to duration of feeding inhibition and energy expenditure (207–208). Moreover, the two peptides appear to stimulate different areas of the brain, with GLP-1 activating neurons in both the hypothalamus and brainstem, and OXM activating neurons exclusively in the ARC nucleus (206). Thus, although the hypothalamic GLP-1 receptors appear to mediate OXM effects, it is likely that the two peptides use different CNS output pathways to achieve their effects (196, 206). Nevertheless, chronic subcutaneous administration of OXM to obese humans significantly reduces calorie intake and body weight and enhances energy expenditure (209–210), suggesting a role for OXM in human weight-loss therapy.
Ghrelin
Ghrelin is a 28-amino acid peptide that holds the unique status of being the only circulating orexigenic peptide of peripheral origin described to date. Ghrelin, which binds to the growth-hormone-secretagogue receptor (GHS-R), is expressed in and released into the circulation by X/A-like cells of the stomach mucosa (211), and crosses the blood–brain barrier (BBB) into the brain (212). Circulating ghrelin levels increase during fasting and between meals in humans and decrease following feeding (213). Central and peripheral administration of ghrelin in rodents and peripheral administration in humans acutely stimulates feeding, while chronic administration in rats causes hyperphagia and obesity (214–216). These data strongly suggest that ghrelin stimulates hunger and is involved in initiating meals (217). Ghrelin is believed to act directly in the hypothalamus by crossing the BBB and binding to GHS-R on NPY-AgRP neurons in the ARC, stimulating the expression of NPY (216, 218). In support of this notion is the fact that ghrelin does not stimulate food intake in NPY and AgRP knockout mice (219). In contrast, there is evidence that the vagus nerve actually mediates the orexigenic effects of ghrelin, but this notion remains controversial, with both positive (220–221) and negative (222) effects reported. Further complicating the issue is the fact that ghrelin-expressing neurons have also been identified in the hypothalamus (223). Thus, the role of peripheral vs. central signaling of feeding stimulation by ghrelin remains to be clarified. It is clear, however, that manipulations of ghrelin or GHS-R expression or function alter feeding and body weight. Antagonists to both ghrelin itself and GHS-R have been found to inhibit food intake and reduce obesity (224–225). Moreover, use of ghrelin antagonists and GHS-R knockout models has led to the hypothesis that ghrelin plays a longer-term role in the regulation of body fat independent of stimulation of food intake (226–227). It is important to note that obese humans have decreased ghrelin levels (228), suggesting that ghrelin may not be involved in the maintenance of obesity. This observation also indicates, however, that ghrelin antagonists may have limited efficacy as weight-loss agents.
Apo A-IV, Enterostatin, and Bombesin-Like Peptides
There are several additional gastrointestinal factors that inhibit feeding when administered systemically or centrally, but whose roles in the control of feeding are not yet clearly defined. The first of these is apolipoprotein A-IV (Apo A-IV), a glycoprotein incorporated into lipoproteins and chylomicrons in response to fat ingestion, and which is released from these particles when they are hydrolyzed by lipoprotein lipase (LPL) in the periphery (229). Initially, intravenously administered Apo A-IV was found to decrease meal size, calorie intake and weight gain in rats (230). Shortly thereafter, central administration of the glycoprotein was shown to have the same effects at considerably smaller doses, and the hypothesis was advanced that Apo A-IV or a fragment of it crosses the BBB and acts at the hypothalamus to inhibit feeding (231). It has since been shown that Apo A-IV is expressed in the hypothalamus and is regulated by leptin (232–233). Moreover, hypothalamic Apo A-IV is downregulated in rats fed a high-fat diet, raising the possibility that this mechanism could contribute to diet-induced obesity (234). At issue is whether Apo A-IV can act quickly enough and is produced in sufficient quantities following meal ingestion to act as a peripheral short-term satiety factor (235). Indeed, a recent report indicates that peripheral Apo A-IV does not cross the BBB (236). It may be more likely that this glycoprotein plays a role in the long-term regulation of calorie intake and body weight via central regulation of its expression by leptin (233) and other regulatory neuropeptides (236).
Enterostatin is a 5-amino acid peptide cleaved from procolipase which is secreted from the exocrine pancreas in response to fat ingestion (237). Peripheral and central administration of enterostatin specifically decreases dietary fat intake in experimental animals (238–239), and these effects are blocked by vagatomy (240). This observation, plus the finding that peripheral enterostatin activates neurons in the NTS, indicates that afferent information triggered by enterostatin is carried to the hindbrain via the vagus nerve and initiates the satiety response (240). However, the central mechanisms underlying enterostatin’s feeding-inhibitory action appear to be complex, with 5-HT1B serotonin receptors and the melanocortin system in the hypothalamus mediating the peptide’s effects (241–242). Moreover, procolipase and enterostatin gene expression have been found in several areas of the rat brain, including the PVN and the ARC (243). Maintenance on a high-fat diet is required to induce the feeding- and body weight-inhibitory effects of enterostatin in rats (244–245), which would suggest a possible role for the peptide in obesity treatment. However, enterostatin administration to humans has to date shown no feeding, energy expenditure, or body weight-altering effects (246–247).
Bombesin is a small peptide originally isolated from amphibian skin. Its closely related mammalian homologs, gastrin-releasing peptide (GRP) and neuromedin B (NMB), are expressed in the stomach and throughout the gastrointestinal tract, and effectively reduce feeding in animals and humans under experimental conditions (248–250). Bombesin and its related peptides inhibit feeding when administered centrally or peripherally, and a variety of receptors appear to mediate their effects (251–252). Unlike the effects of CCK, the feeding-inhibitory effects of bombesin-related peptides are not dependent on an intact vagus nerve (253), and the exact mechanism underlying their satiety action remains unclear. Also uncertain is whether these peptides are regulated by ingested nutrients under normal feeding conditions (196). Of the bombesin-like peptide receptors, knockout of the bombesin receptor subtype-3 (BRS-3) in mice results in hyperphagia, obesity, and a reduced metabolic rate (254). Conversely, long-term pharmacological stimulation of BRS-3 in mice increased metabolic rate and reduced food intake and body weight (255), demonstrating the potential involvement of this class of receptors in energy homeostasis.
Pancreatic Peptides and Liver Metabolic Signals
Amylin and Glucagon
There are two additional peptides released by the pancreas involved in the control of food intake and body weight. We have already described the role of insulin. Amylin is a 37-amino acid peptide co-secreted with insulin from the β-cells of the pancreas in response to a meal. In addition to its glucoregulatory effects, which include the inhibition of gastric emptying (256), amylin inhibits food intake by reducing meal size without increasing meal frequency (257–258) and interacts synergistically with CCK in its satiety effects (259). Amylin binds to the AP in the hindbrain and also modulates neuronal activity in the hypothalamus and nucleus accumbens, a reward-related brain site (260–261). Thus, it is believed to be involved not only in the integration of peripheral meal-related signals, but also in the long-term regulation of body weight (22). Consistent with this hypothesis are findings that microinjection of low doses of amylin into the 3rd ventricle adjacent to the hypothalamus potently reduces feeding (262), and blockade of central amylin receptors increase food intake, body weight, and body fat (263). More recently, it was reported that the feeding inhibitory effects of amylin are also mediated by activation of the AP in the hindbrain (264). The putative dual role of amylin in feeding and body weight regulation may indicate a therapeutic role for this peptide, since chronic administration of amylin to obese humans and rats has been shown to reduce body weight (265–266).
Glucagon is a 29-amino acid peptide secreted from the α-cells of the pancreas in response to both oral nutrient stimulation and the ingestion of meals (267–268). Release of glucagon from the pancreas during a meal results in elevated enteric levels of the peptide, which act in the liver to stimulate hepatic glycogenolysis (269). This process in turn results in feeding inhibition via reduced meal size, but not meal number (270–271). The satiety process initiated by glucagon is mediated by the vagus nerve; vagatomy and lesions of the afferent receptor fields in the NTS eliminate the response (272–273). Like amylin, glucagon appears to act synergistically with CCK to inhibit feeding (274). Glucagon-induced satiation appears to be a physiological process, since administration of glucagon antibodies to rats increases meal size (271, 275), while administration of glucagon to humans reduces meal size and is accompanied by feelings of satiety (276). However, the hepatic and central mechanisms underlying glucagon’s feeding-inhibitory effects have not been identified (22), although a response to increased nutrient availability in the liver clearly is implied (see following section).
Liver Energy Status
It has been known for some time that systemic administration of inhibitors of glucose and fatty acid oxidation stimulates feeding (277–278). While this response may in part be mediated by nutrient-sensing mechanisms in the hypothalamus (addressed in the next section), the hypothesis has been advanced that this response also occurs in the liver and is based on liver energy status (279). Specifically, feeding stimulated by inhibition of hepatic glycolysis and/or fatty acid oxidation may be triggered by a depletion of intracellular hepatic ATP, via an alteration in the ATP/ADP ratio and a reduction in intracellular phosphorylation potential (279–280). A considerable volume of evidence has accumulated in support of this hypothesis. First, agents that interrupt glucose utilization and fatty acid oxidation specifically in liver act synergistically to increase food intake while decreasing the hepatic ATP/ADP ratio (281–282). Second, a reduction in and recovery of markers of liver energy status have been found to closely track the course of imposed food deprivation and subsequent refeeding in rats (283). Finally, a study in diabetic rats that are not able to use glucose, and therefore are hyperphagic, has demonstrated that high-fat diet feeding eliminates diabetic hyperphagia and normalizes elevated hypothalamic levels of NPY and CRF. Crucial to the assertion that this response is based in the liver is the observation that severing of the hepatic branch of the vagus nerve eliminates the high-fat diet-induced feeding effect and the normalization of NPY and CRF levels in the diabetic animals (284). Although the precise mechanism whereby alterations of hepatic ATP levels signal liver energy status to the brain remains unknown, exploratory work has identified sodium pump activity and intracellular calcium release as potentially involved in this process (285–286). A recent study in rats with an inherited susceptibility to diet-induced obesity demonstrated the decreased expression of hepatic enzymes involved in fatty acid oxidation, including carnitine palmitoyl transferase-1 (CPT-1), the enzyme that mediates the transport of long chain fatty acids into mitochondria (287). Thus impairments in hepatic energy processing may be involved in a predisposition to dietary-induced obesity.
Brain Nutrient Sensing
In addition to responding to the myriad of central and peripheral peptide signals described above, the brain is sensitive to nutrient-based intracellular signals and also uses these factors in the control of food intake. This is a complex process in which all three major fuel substrates appear to be involved and which has only recently come under intense investigation. We will start our discussion by examining the role of adenosine monophosphate-activated protein kinase (AMPK), an intracellular enzyme that, because of its involvement in basic cellular energy homeostasis, may represent the final common pathway for these nutrient-based feeding-altering effects.
Role of AMP Kinase
AMPK was originally identified as a regulator of cellular energy balance, becoming activated by a decrease in intracellular energy reserves via an increase in the AMP/ATP ratio. Activation of AMPK in turn stimulates substrate uptake and oxidation pathways, which eventually generate additional ATP and restore adequate levels of intracellular energy reserves (288). More recently, the role of AMPK was broadened by the observation that it serves as a fuel gauge, determining whether fat is oxidized or stored in muscle (289). Accumulating evidence now indicates that changes in hypothalamic AMPK activity are also involved in the control of feeding, reflecting both nutrient levels and hormonal signals from the periphery (290–291). Thus, activation of AMPK in response to nutrient insufficiency stimulates substrate oxidation and increases feeding, while inhibition of its activity in response to nutrient surfeit decreases substrate oxidation and inhibits food intake (288) (see Figure 8-6). It has now been demonstrated that AMPK activity is inhibited in the ARC, PVN, and other hypothalamic regions by leptin, glucose, and insulin, and is responsive to fasting and refeeding with appropriate increases and decreases, respectively (291). It should be noted that leptin has the opposite effect on AMPK activity in muscle tissue (289, 292). The substrate oxidative and feeding effects of hypothalamic AMPK appear to be mediated by two mechanisms: 1) AMPK-induced alterations in the fatty acid synthetic pathway that alter intracellular malonyl-CoA levels, which in turn determine the degree of long-chain fatty acid oxidation taking place in the mitochondria (see section on Fatty Acids below); and 2) AMPK-mediated alterations in expression levels of NPY and AgRP in ARC neurons, which directly stimulate or reduce feeding (291, 293). In vivo effects of AMPK manipulations have been shown in rodents. Central administration of α-lipoic acid, an inhibitor of AMPK activity, leads to reduced food intake and body weight in rats (294), while expression of constitutively active AMPK in the hypothalamus of mice leads to increased feeding and weight gain (291). Alterations of AMPK activity, as we shall see, are hypothesized (288, 295) to be involved in all forms of brain nutrient sensing and subsequent feeding effects (Figure 8-6).
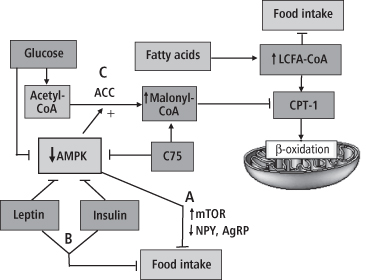
Figure 8-6 Hypothesized central role of AMP-activated protein kinase (AMPK) in nutrient-based feeding effects. The activity of AMPK, a sensor of intracellular fuel levels, is decreased by glucose, leptin, insulin, and the fatty acid synthesis inhibitor C-75. Decreases of AMPK activity, and central administration of the amino acid leucine, reportedly act at (A) to stimulate mTOR activity and reduce the expression of NPY and AgRP in ARC, thus inhibiting feeding. Leptin and insulin may also act to reduce feeding at (B) via the melanocortin pathway, not directly linked to AMPK activity. Decreases of AMPK activity also dephosphorylate and activate the enzyme acetyl co-A carboxylase (ACC) at (C), which elevates malonyl Co-A levels, which in turn inhibit the enzyme carnitine palmityoltransferase-1 (CPT-1), reducing the flow of long chain fatty acyl Co-A (LCFA-CoA) into the mitochondria for β-oxidation. This enlarges the intracellular LCFA-CoA pool, which, in an as yet unidentified manner, is hypothesized to inhibit feeding. Note that central administration of oleic acid, a LCFA, enhances the LCFA-CoA pool and inhibits feeding. The reverse processes takes place when intracellular fuel levels or feeding-inhibitory hormone levels are decreased, resulting in activation of AMPK, elevations of NPY and AgRP, increases in CPT-1, decreases in the LCFA-CoA pool, and feeding stimulation.
(Adapted and reprinted with permission from Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nat Neurosci 2005; 8: 579–84.)
Glucose
At first glance, glucose would appear to be the ideal nutrient signal of brain and peripheral energy status, since it is the basic fuel of the body. Indeed, one of the very first theories of control of feeding was the “glucostatic hypothesis,” which proposed that increases and decreases of circulating glucose, detected by the brain, served as satiety and hunger signals, respectively (296). In support of this notion was the demonstration that 3rd ventricular infusions of glucose inhibit feeding (297), while infusions of glucosamine, an inhibitor of the glucose-transporting enzyme glucokinase, into the same brain site stimulate feeding (298). However, numerous investigators were unable to demonstrate an effect of acute peripheral infusions of glucose on meal taking (299–301), and the glucostatic hypothesis in its original form has been abandoned. The well-known feeding-stimulatory effects of systemic hypoglycemic doses of insulin (302–303), and of 2-deoxyglucose (2-DG), an inhibitor of glucose transport and utilization (278), are now viewed as “emergency” responses to acute cellular glucoprivation, and not the expression of normal energy homeostatic pathways (304).
The notion of brain glucose sensing playing an integral role in energy homeostasis has been revived recently, with the discovery of glucosensing neurons in the hypothalamus, including in NPY and POMC expressing neurons in ARC, which are also responsive to leptin, insulin, and other neuropeptides (304–305). It has been hypothesized that glucose, insulin, and leptin act in such neurons via glucokinase and phosphatidylinositol-3-OH kinase (PI3-kinase)-mediated second messenger pathways, respectively, to stimulate intracellular glucose metabolism, which provides ATP and in turn reduces AMPK activity and feeding (304, 306). Reductions of glucose sensing in such neurons brought about by inhibition of the glucose transporter and receptor GLUT-2 (307), and by high fat feeding (308), stimulate food intake, and may lead to obesity and the development of type 2 diabetes (306). Note that the above hypothesis switches the emphasis of glucose’s role in energy homeostasis from short-term, meal-related effects to long-term energy balance adjustments. In support of this notion, it has been shown that chronic, as opposed to acute, systemic infusions of glucose reduce food intake and body weight in both lean and obese rats (309–310). Moreover, chronic administration of 5-thioglucose, another inhibitor of GLUT-2, into the 4th ventricle of rats stimulates acute hyperphagia and body fat gain (311). Considerably more needs to be learned about the effects on feeding and body weight of glucose action in the hypothalamus before we can fully appreciate its role in the control of energy balance.
Fatty Acids
Recently, 3rd ventricular administration of oleic acid was shown to reduce food intake and peripheral glucose production in rats (312). This was the first demonstration that long chain fatty acids (LCFA) delivered centrally can inhibit feeding, although central administration of both glycerol and ketones has long been known to have this effect (313). This finding stimulated a spate of research in the area of CNS lipid sensing, generating further data indicating that the effect is based on the intracellular accumulation of LCFA-CoA, the esterified form of LCFAs, normally transferred into the mitochondria by the enzyme CPT-1, for β-oxidation (314). Thus, the accumulation of LCFA-CoA is hypothesized to inhibit feeding, while depletion of the LCFA-CoA pool by its entry into the mitochondria and subsequent oxidation would act to stimulate feeding. In support of this notion, inhibition of hypothalamic CPT-1 was found to decrease food intake in normal rats (315) and to normalize food intake in high-fat-fed rats, which become unresponsive to central infusions of oleic acid (316–317). Moreover, central oleic acid administration was found to inhibit the expression of both NPY and AgRP in the hypothalamus (316). No specific mechanism for the mediation of LCFA-CoA pool effects on feeding has yet been identified, however.
Crucial to the hypothesized mechanism of LCFA-CoA signaling is the production of intracellular malonyl-CoA, which inhibits CPT-1 as its levels increase (314). Inhibition of CPT-1 reduces mitochondrial fatty acid oxidation and thereby protects the LCFA-CoA pool. As noted above, AMPK activity is a major determinant of the level of hypothalamic malonyl-CoA, decreasing it during fasting and increasing it in the fed (energy-replete) state, via deactivation and activation, respectively, of the malonyl-CoA-generating enzyme acetyl-CoA carboxylase (ACC) in the fatty acid synthetic pathway (293). Thus, AMPK activity is crucial to expression of LCFA-CoA feeding effects (shown in Figure 8-6). Note that glucose is also a major determinant of AMPK activity and thus of malonyl-CoA production, acting to decrease AMPK activity presumably by providing intracellular ATP (318). Support for the role of malonyl-CoA in lipid sensing can be found in the demonstration that hypothalamic overexpression of malonyl-CoA decarboxylase, the enzyme that degrades malonyl-CoA, leads to hyperphagia and obesity in rats (319).
Additional support that alterations of CNS intracellular fatty acid metabolism are involved in the control of feeding is seen in the effects of C75, a synthetic inhibitor of fatty acid synthesis which elevates intracellular ATP levels, thereby decreasing AMPK activity and inhibiting feeding (320–321). This action of C75 presumably increases the intracellular LCFA pool by increasing hypothalamic malonyl-CoA levels, which in turn inhibit CPT-1 activity (322–323). Centrally administered C75 blocks fasting-induced increases of hypothalamic NPY and AgRP (324), and decreases food intake and body weight when administered centrally or peripherally to rats (320, 325). Central administration of C75 also rapidly inhibits ghrelin secretion from both hypothalamic tissue and the stomach mucosa (326). It is unclear whether decreased CPT-1 activity is consistently associated with the feeding effects of C75, since newer research has found that central C75-induced elevations of CPT-1 activity may be associated with its anorexic effects (327). Thus, although the exact mechanism by which CNS intracellular fatty acid metabolism alters feeding remains controversial, it is clear that this metabolic pathway plays a significant role in the nutrient-based control of energy balance.
Amino Acids
Recently, evidence has emerged that representatives of the third major nutrient class, amino acids, may also be sensed in the hypothalamus and participate in the control of food intake. The mammalian Target of Rapamycin (mTOR) is an intracellular signaling molecule involved in protein synthesis and cell growth throughout the body, which is stimulated by both hormones and nutrients (328–329). A recent study in rats identified the presence of mTOR and its second messenger components in ARC NPY and AgRP neurons, and demonstrated that their expression is regulated by fasting and refeeding (330). The investigators went on to show that 3rd ventricular administration of the branched-chain amino acid leucine activates mTOR, decreases the expression of ARC NPY and inhibits feeding. Moreover, the feeding inhibitory effects of leucine, and of the hormone leptin, were blocked by administration of the potent mTOR inhibitor, rapamycin. The feeding-inhibitory effects of melanocortin receptor agonists were unaffected by rapamycin, however (330). Thus, the mTOR pathway appears to mediate the feeding inhibitory effects not only of the amino acid leucine, but those of leptin that are specifically dependent on NPY expression. A second study has replicated the leucine-mTOR feeding-inhibitory effect and further demonstrated that mTOR activation also inhibits AgRP expression levels in hypothalamic neurons (331). In this second study, an amino acid mixture containing leucine had a feeding-inhibitory effect equal to the same concentration of leucine administered alone, implicating leucine as the key amino acid signal for brain sensing. These data are consistent with enhanced mTOR activity and weight loss recently observed in lean rats and obese mice fed a leucine-containing high-protein diet (332). Importantly, earlier work has demonstrated that mTOR activity itself is decreased by AMPK activation (333), which suggests that reductions in AMPK activation in the hypothalamus may also induce mTOR feeding-inhibitory activity (332, 334). This is consistent with the finding that decreased AMPK activation is essential for expression of the feeding-inhibitory effects of leptin (291). Thus, alterations in AMPK activation may regulate both the leptin NPY/AgRP and the leucine-mTOR feeding inhibitory pathways (see Figure 8-6). It is clear that the complex interactions between intracellular fuel levels, their utilization, and their effects on neurotransmitter expression and feeding are only beginning to be understood.
Adipose Tissue Influences
The role of leptin and other hormones secreted from adipose tissue in the regulation of energy balance has been well described (335–336). Even apart from its endocrine and metabolic effects, however, adipose tissue remains an extraordinary organ, playing a remarkable additional role in overall energy balance through its ability to continuously proliferate adipocytes when provided with sufficient substrate, and to actively regulate its own mass. We will consider adipocyte proliferation first.
Adipocyte Proliferation
It has long been evident that the total number of adipocytes in the body may increase under appropriate dietary conditions, including the availability of highly palatable, high-energy diets (337). This process is believed to involve first the enlargement of adipocytes through lipid filling, and then as the cells approach maximal or “peak” size, the initiation of adipocyte proliferation, which increases the body’s total adipocyte number (338–339). The result is, of course, elevated body weight and body fat as the new cells become replete with lipid. Because mature adipocytes cannot be eliminated in any way except surgically, a new permanent body weight will be attained. Moreover, the tendency for adipocytes to maintain their replete size via hormonal (336) and metabolic (340–341) mechanisms means that the new, elevated body weight will be defended in the face of calorie restriction and weight loss. Thus, the simple ability of adipose tissue to proliferate its own cells can elevate energy homeostatic requirements and “reset” the body weight maintenance range to higher levels. Although our knowledge of the proliferative process involved in diet-induced adipocyte number increases is limited, evidence indicates that the duration and severity of the obesity induced may determine whether or not this process is reversible (342).
Regulation of Adipose Mass
It is well known that the surgical removal of adipose tissue (lipectomy) from specific fat depots in rodents, particularly during growth, is followed by restoration of the depleted mass in other depots as the animals mature (343). Restoration of the depleted fat tissue is quite precise, indicating that the body has the remarkable ability to monitor its own total fat mass. This phenomenon has recently been systematically studied in human subjects, and results are consistent with the notion of adipose mass regulation in humans as well (344). An understanding of the mechanisms underlying adipose mass regulation has been lacking until relatively recently, when an impressive series of studies appeared demonstrating the sympathetic innervation of white adipose tissue (345). Further work has shown that sympathetic innervation is an important regulator of adipose tissue protein secretion, adipocyte lipolysis, and adipocyte proliferation (346). Indeed, the removal of sympathetic stimulation from white adipose tissue via surgical or toxin-induced denervation stimulates adipocyte proliferation (347). Further, it has been shown that sensory innervation of white adipose tissue exists (348), and via a feedback loop to the CNS probably modulates sympathetic tone and thereby adipocyte lipolysis (345). Importantly, sensory nerve denervation has the effect of mimicking the compensatory increases in adipose tissue mass that typically follow lipectomy (349). Thus, it appears that neural sympathetic and sensory innervation of white adipose tissue play a hitherto unsuspected but significant role in the regulation of adipose tissue mass. Whether and how these mechanisms are involved in diet-induced increases of adiposity remain to be investigated, as does the nature of the CNS process which itself controls total fat mass.
Characteristics of the Weight Regulatory System
Defense of Elevated Body Weight
It is clear that the regulation of feeding and body weight is determined by multiple systems, with overlapping neural, endocrine, and metabolic mechanisms designed to maintain short- and long-term energy reserves at relatively constant levels. Obviously, these mechanisms not only interact with each other, but appear to compensate for each other in the face of interventions that alter their functioning. Thus, minimal or no effects on feeding or body weight under standard feeding conditions are seen in animals with knockouts of the NPY, AgRP, or ghrelin genes, three major players in the energy homeostatic process (226, 350). Undoubtedly, this is one of the characteristics of the regulatory system that renders appetite and body weight reduction so difficult over the long term. Moreover, while energy regulatory mechanisms have been demonstrated that respond to both decreases and increases of body weight, the energy homeostatic system appears to respond more vigorously to weight loss than to gradual weight gain (351). Among these responses is a reduction of resting and total energy expenditure following weight loss, which reduces the total calorie requirements of the body and increases the likelihood of excess calories being stored if they are consumed during attempted weight maintenance (352–353). Finally, as we saw above, in response to calorie overconsumption, adipocyte number and thus total body fat can permanently increase in the adult state via a proliferative process that appears largely unopposed and results in higher body weight. If the new body weight persists over a long enough period, it will readily be defended by the body weight regulatory system.
What Is Disordered in Obesity?
The sheer number of energy homeostatic signals described above indicates that there may be numerous candidates for alterations in the regulatory system that may result in the onset and/or maintenance of obesity. We will briefly consider four categories of factors that may shift energy balance toward the development of obesity: genetic, developmental, environmental, and hormonal. It is likely that alterations in these factors are not mutually exclusive, and in practice they are likely to interact in the development and/or maintenance of obesity.
Genetic Factors
The most obvious of the genetic alterations involved in excess body weight are those arising from monogenic (single gene) mutations, best represented by mutations in the leptin and melanocortin systems. These result in severe early-onset obesity, but such conditions are rare (354). Potentially more subtle monogenic mutations that may underlie the gradual onset of obesity have recently been identified experimentally (175, 355) and in human studies (356–358). More common but less well defined are “polygenic” factors, represented by alterations in multiple genes that together enhance the susceptibility to obesity (359). Numerous candidate genes and chromosome markers of this type have been identified recently in human genome-wide association scans (360–362). In most cases, the function of candidate genes identified in this way remains to be determined, as does their potential interaction in inducing and/or maintaining increased susceptibility to obesity. The role of gene polymorphisms and gene interactions in obesity and associated metabolic disorders is now under study by numerous investigators, although these are complex molecular processes (363).
Developmental Effects
A second factor involves developmental programming in offspring of brain pathways that result in alterations of the energy regulatory system, which predispose to obesity. Recent research has demonstrated the existence of critical periods in utero and during early postnatal growth during which the development of neural circuits and metabolic pathways can be modified, resulting in elevated feeding behavior and body weight gain later in life (364). For example, leptin is a crucial player in early hypothalamic pathway development, exerting potentially protective anti-obesity effects on hypothalamic circuitry during gestation and in the early neonatal period (365–367). While leptin’s early effects normally are protective, studies have identified conditions under which leptin and insulin can exert detrimental effects related to the onset of obesity (368–369). The extent to which such perinatal developmental influences can result in permanent alterations of the brain’s energy regulatory system is now the focus of intense research interest (370). On a more practical note, numerous experimental studies have demonstrated obesity- and diabetes-inducing effects on offspring of elevations in maternal body weight and/or alterations in diet composition during pregnancy (371–372). Such findings have stimulated numerous studies on potential correlations between human maternal body weight and/or gestational weight gain, and the feeding and body weight characteristics of the offspring (373–374). These studies indicate that maternal factors relating to body weight appear to have significant effects on the development of obesity seen in children (see Chapter 10).
Environmental Influences
The third factor to be considered, while not qualifying as a disorder, is now viewed as a major risk factor for the development of obesity. This is the abundant availability of food and reduced requirements for activity that characterize modern environments (375). As noted above, biological mechanisms designed to protect us against excess weight gain are not as vigorous as those that protect us against weight loss (351). The result of these environmental alterations may well be the accumulation of excess body fat by a large proportion of the population. The effects of an abundant, calorie-dense food supply on caloric intake and body weight have been amply demonstrated in rodents (376) and have led to the hypothesis that some individuals within a population are more susceptible than others to the availability and effects of appetizing food items (7). This notion has its origin in the “thrifty gene” hypothesis, which states that as mammals evolved, the unreliability of their food sources rendered excess calorie intake and the accumulation of body fat an advantage for survival (377–378). Although thrifty genes have yet to be discovered, obesity-prone animals are readily identified by their tendency to over-consume a palatable diet at an early age (379), and attempts currently are underway to identify characteristics that reliably predict the subsequent development of obesity (380–381). Similar attempts at linking diet susceptibility to specific genes are also being made in humans (382). Reductions of physical activity by individuals in the current environment would further augment the above weight gain-inducing tendencies of susceptible individuals (383–384).
Hormone Resistance
In addition to the above influences on energy homeostasis that predispose to obesity, there remain potential alterations of the very hormonal mechanisms involved in weight regulation. These include the onset in wild-type animals exposed to high-energy diets of resistance to leptin- and insulin-mediated hypothalamic signaling, which become significant as obesity develops and levels of these hormones rise (385–386). Resistance to the feeding-inhibitory effects of both leptin and insulin has been well documented experimentally (387–388) and is believed to play a role in the maintenance and, in some cases, onset of obesity (389–391). Several potential mechanisms mediate leptin and insulin resistance including alterations of blood–brain barrier hormone transport (392–393), hypothalamic hormone receptor expression (394–395), and receptor second-messenger signaling (396–397). It should be noted that leptin resistance may occur in peripheral tissue as well (398–399). What makes hormone resistance troublesome is the fact that it is expressed not only in animals with well-defined mutations of the leptin and insulin systems, but in wild-type populations with no known genetic alterations. Thus, leptin resistance can readily be induced in laboratory rodents by dietary means (400) and is seen in both obesity-resistant and obesity-prone rat strains (401–402). Although leptin resistance can rapidly be induced with a high-fat diet (403), there is evidence that the process is reversible with diet modification and weight reduction (404). Indeed, the reinstatement of leptin sensitivity following significant weight loss may offer a rationale for leptin treatment of weight-reduced individuals (405–406).
Implications for Treatment
The characteristics of the energy balance system that so effectively protects against body weight loss are now being addressed by workers attempting to design effective weight reduction approaches (375). However, the complexity of the regulatory system makes this task daunting. New concepts in the design of anti-obesity drugs include the targeting of peripheral peptides and their receptor systems in the CNS (407–408), the development of effective combinations of feeding-inhibitory peptides for treatment (409–410), and interventions based on metabolic and adipose tissue factors involved in energy balance (411–412). The use of knockout and transgenic methods for the study of appetite and body weight regulation undoubtedly will aid in the design of novel weight loss agents (413). Along with these pharmacological and molecular approaches is a new awareness of the interaction between energy homeostatic systems and the food environment we are exposed to in modern society (414). This awareness has already led to environmental modifications designed to reduce calorie intake and increase energy expenditure. Thus, the next decade holds great promise for the development of effective therapeutic advances based on these new approaches.
Summary: Key Points
- The energy balance equation shows the relation between energy intake, expenditure, and stores as:
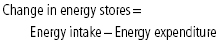
- “Energy homeostasis” refers to the collection of biological phenomena involved in the complex regulation of energy intake and energy expenditure to maintain constant energy stores (and hence body weight).
- In the simplified model of physiological appetite–body weight regulation, there are short- and a long-term components. Integration of long- and short-term signals takes place in two hypothalamic areas and one hindbrain center.
- The short-term component of the model involves hindbrain control of the onset and cessation of feeding on a meal-by-meal basis and is responsive to a variety of signals from the body.
- The long-term component of the model monitors and regulates body nutrient stores over weeks or months, integrating hormonal signals from the body’s fat mass (i.e., energy stores). The integrative hypothalamic centers also coordinate input from and to the hindbrain.
- A wide array of peptide,hormone, and neurotransmitter signals influence energy homeostasis. These can affect feeding by altering hunger or motivation to eat; satiety; macronutrient appetite or intake; responsiveness to various properties of food; meal size, frequency, or duration; the between meal or inter-meal interval; and responsiveness to gastric distension.
- Non-homeostatic reward systems, which include the dopamine, opioid, and endocannabinoid systems, are capable of overriding physiological homeostatic feeding systems to promote overeating of food even in the absence of hunger.
Related posts:
 The application of public health lessons to stemming the obesity epidemic
Impact of obesity during adulthood on chronic disease: diabetes, hypertension, metabolic syndrome, cardiovascular disease, and cancer
The application of public health lessons to stemming the obesity epidemic
Impact of obesity during adulthood on chronic disease: diabetes, hypertension, metabolic syndrome, cardiovascular disease, and cancer
 Eating behaviors in obesity: the role of appetite
Eating behaviors in obesity: the role of appetite
 Treatment of overweight and obesity in children
Treatment of overweight and obesity in children
 Behavioral approaches to the treatment of obesity
Behavioral approaches to the treatment of obesity
 School-based interventions for overweight and obesity prevention
School-based interventions for overweight and obesity prevention



