Fig. 7.1
Anatomy of the cerebral blood supply. (a) Circle of Willis. (b) The arterial supply of the deep white matter arises from branches of the ACA and the MCA. The supply of the basal ganglia white matter is provided by arteries arising directly from the circle of Willis and its proximal branches. (c) Anatomy of the wall of arteries, arterioles, and capillaries. ACA anterior cerebral artery, ICA internal carotid artery, MCA middle cerebral artery, PCA posterior cerebral artery. Reproduced with permission from [1]
Because of the structure of the arteries and their relation to the environment of the brain, especially due to the Virchow–Robin space, it is only at the level of the neurovascular unit that initiation of the match between the neuronal metabolic needs and the blood flow can be regulated. This supposes, therefore, a singular mechanism of retrograde communications from arterioles and capillaries of the metabolic domain (the neurovascular units) to the resistance domain (pial arteries and arterioles) that integrate upstream systemic pressure and carotid flow with downstream neuronal metabolic demand.
Finally, it is worth mentioning that brain vascular resistance is very low. Thus pulsation pressure extends in brain and is transmitted through the capillary network to the veins [37]. In consequences, any increases in pulse pressure may lead to brain damage, the so-called pulse wave encephalopathy.
2.2 The Coordinated Cerebrovascular Function
From the previous chapters, you will already know that the metabolic needs of the brain are important while it has no energy reserve [32, 38]; simple mechanical whisker stimulation immediately increases cortical blood flow by 30 % in 3-month-old mice [39], highlighting the necessity of a close match between energy demand and blood flow. Mismatch between the two could create astrocyte and neuronal stress and threatens their survival.
In resistance arteries, essentially the pial arteries, the integration of the endothelial shear stress signals (flow-dependent tone) and the vascular smooth muscle cells (VSMC) pressure signals (myogenic tone) is required to achieve adequate blood supply to the capillaries. Activated neurons, either directly or using astrocytes as relays, stimulate nearby arteriolar smooth muscle cells to induce their dilation [30, 32, 40]. Dilatory mediators are numerous, including the neurotransmitters themselves (neuron-derived nitric oxide (NO)), astrocyte-derived mediators (NO, prostaglandin E2, epoxyeicosatrienoic acids (EETs), potassium ions (K+)) as well as metabolites (lactate, adenosine). Hence, the endothelium is not involved in the initial and local dilatory response evoked by neuronal metabolic demands. If sustained, however, this initial dilatory response will create a local drop in resistance necessary to create the driving force for blood flow with the retrograde conduction of vasodilatation (RCVD).
Two principal endothelium-dependent mechanisms identified in vitro in cerebral arteries are responsible for the RCVD: (1) the transmission of chemical and electrical signals through gap junctions connecting adjacent endothelial cells (EC) and VSMC (i.e., myo-endothelial junctions) [41–45] and (2) flow-mediated dilation (FMD) [43, 46, 47].
Longitudinal spreading of a hyperpolarizing current initiated by activation of local receptor- or channel-mediated membrane hyperpolarization could mediate cerebral RCVD [41, 42]. The increased expression level of gap junctions within the endothelium [44] and inhibition of the conducted dilatory response by endothelial impairment suggest that the endothelium is the favored conduction pathway in brain arteries [42, 48]. A local RCVD at the level of penetrating arterioles would magnify the drop in resistance induced by neurovascular coupling and increase shear stress in the upstream pial arteries [49]. An acute experimental increase in blood flow in vivo leads to a transient increase in shear stress that triggers remote vascular FMD [46].
Shear stress (dyn/cm2), the frictional force per unit area acting on the inner vessel wall, is a function of flow rate, blood viscosity, and the third power of the inside radius of the artery [50–52]. Shear stress is relatively constant from the aorta to the capillaries varying only between 10.4 and 26 dyn/cm2 [50, 53], averaging 15 dyn/cm2. Any local chronic [54] or acute [55] variations in blood flow will alter shear stress that will trigger a change in arterial diameter aiming at normalizing shear stress level. Indeed, the surface of the vascular endothelium is structurally organized to detect wall shear stress variations [56] leading to the release of several endothelium-derived relaxing factors (EDRFs) [57]. Cerebral FMD is abrogated in vitro after endothelium removal [55, 58, 59]. Nitric oxide produced by eNOS and additional EDRFs generate the flow response [55, 60, 61]. Hydrogen peroxide, produced after eNOS [55] and nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase [62] activation, contributes as well to shear-dependent dilation of rodent cerebral arteries, but not in all cases [61]. Partial abolition of FMD by inhibitors of eNOS [55] or NAPDH-oxidase [62] implies the participation of yet other EDRFs.
Flow-mediated constriction (FMC), largely endothelium-independent, also develops in isolated human cerebral arteries [63]. FMC was reported in isolated rat cerebral arteries as well [64, 65], while in isolated cat middle cerebral arteries, FMC was accompanied by smooth muscle cell membrane depolarization [66]. In piglet, however, low flow rates induced a constriction of pressurized cerebral arteries while high flow rates induced dilation, both responses being dependent of an intact endothelium [67]. Likewise in rabbit pressurized isolated arteries, a combination of both FMD and FMC has been reported at different flow rates, but only FMD was abolished by endothelium denudation [58, 68]. While FMD and FMC are potential mechanisms that could regulate local CBF on the moment-to-moment basis, the data collected in isolated cerebral arteries prevent us to draw any physiological conclusion. Some of these observations are species-specific rather than cerebral-specific, and vessel size, structure, biomechanics, and initial level of myogenic tone [58, 59] certainly contribute to the experimental outcomes. Furthermore, cerebrovascular shear-dependent responses in humans are very poorly documented [69]. However, an acute change in shear stress is only temporary [70]. Chronically, changes in blood flow in peripheral arteries will unbalance shear stress that will modify gene expression, wall structure, and endothelial function [51, 71] and lead to a new vascular diameter restoring a normal shear stress [71]. This complex response is genetically determined in the mouse carotid artery [72], but its implication in CBF regulation is not known.
3 Pulse Wave Velocity and Brain Damages
3.1 Pulse Wave Encephalopathy
As previously mentioned, brain vascular resistance is very low. Thus small cerebral arterial vessels are exposed to the high-pressure fluctuations that exist in the carotid and cerebral arteries [73]. Any increase in systemic pulse pressure may induce cerebrovascular damage [37]. Indeed, increase in pulsatility and resistance indexes of the mean cerebral artery (MCA), two indexes that reflect downstream increase in vascular resistance, are related to increase in aortic stiffness [74]. Furthermore, arterial stiffening with aging increases carotid flow augmentation that may explain the increase in CBF fluctuations [75]. Several studies establish a link between peripheral arterial stiffness and brain microvascular disease such as cerebral small vessel disease, white matter changes or cognitive function in older individuals [76–78], hypertensive [79, 80] and diabetic patients [81, 82]. More recently, it has been reported that CBF pulsatility is independently associated with central hemodynamics and that higher pulsatility of CBF was positively correlated with the greater total volume of white matter hyperintensity [83]. Finally, leukoaraiosis is strongly associated with cerebral arterial pulsatility that is strongly dependent on aortic pulsatility and large artery stiffness [84]. Thus physiopathological conditions characterized by an increase in peripheral arteries stiffness are associated with brain damages. This may suggest that peripheral arteries stiffness may impact on cerebral arterioles structure and function. It has already been reported that increases in pulse pressure may play a role on cerebral arterioles structure and mechanics [85, 86]. Finally, in a recent study, it was reported that experimental stiffening of the carotid artery of young mice using CaCl2 application, led to collagen synthesis, fragmentation of elastin filaments, leading to an increase in CBF pulsatility and intracranial cerebrovascular damage and neuronal loss [87]. This is the first demonstration that stiffening of the carotid artery alone damages the brain.
3.2 Plasticity of the Cerebrovascular Wall
Cerebral arterioles undergo alteration in wall structure and dynamics as well as in function in conditions associated with an increase in aortic stiffness such as aging, hypertension, and chronic renal failure. However, despite similar impact on aortic stiffness as measured by PWV, structure and dynamics of cerebral arterioles differ in these circumstances. Considering aging, one study revealed that in 24- to 27-month-old Fisher rats, pial arteriolar wall underwent atrophy with the loss of nearly half of the volume occupied by VSMC; the wall became stiffer; the passive internal diameter of deactivated (Ca2+-free experimental conditions) arterioles became smaller [88]. Furthermore, collaterals branching from the MCA and anterior cerebral arteries are altered in old mice, evidenced by a rarefaction collateral number, a reduction in diameter, and an increased tortuosity consistent with a sixfold increment of collateral resistance [89]. In human, it has been reported an association between arterial stiffness evaluated by the brachial-ankle PWV and cerebrovascular resistance in the elderly [90]. Thus, peripheral artery stiffness influences cerebral circulation in this population. However, the study does not allow to determine if the increase in cerebrovascular resistance is consecutive to structural or functional alterations of cerebral arteries and arterioles. However, in another study performed in middle-aged and older individuals, arterial wall stiffening in large arterial beds is associated with retinal arteriolar narrowing indicating that large artery stiffness may impact on cerebral vasculature structure [91]. Postmortem human posterior cerebral arteries (~2.3 mm) were compared in 6 young (~42 years of age) and 6 aged (~70 years of age) adults with severe cardiovascular disorders: aging was associated with a thicker intima and media, a loss of VSMC but an increase in collagen, a severe stiffening and concomitant elastin fiber dysfunction with possible fragmentation and reorganization [92]. In this study however, it may be difficult to differentiate effects of aging from effects of severe cardiovascular disorders.
During chronic hypertension, basilar arteries, posterior cerebral arteries, and the MCA (>200 μm) stiffen in hypertensive rats [93–96]. In contrast, cerebral arteries (>150 μm) and arterioles (<70 μm) distensibility has been reported to increase in rat and mouse models of hypertension [97, 98] and mice deficient in superoxide dismutase [99]. Arterial wall hypertrophy accompanied both hypertension and atherosclerosis and eutrophic inward remodelling was observed in some models of hypertension in rats [100, 101].
Structure and mechanics of cerebral arterioles were not altered in a model of chronic kidney disease in the mice. In contrast endothelium-dependent relaxation was decreased in mice with chronic renal failure [102]. In human, chronic kidney disease is associated with an increase in intracranial arteries calcification. Furthermore calcification of intracranial arteries is associated with a higher risk of stroke and with a greater postischemic stroke mortality and vascular events [103]. However, intracranial arteries calcification is also associated with aortic atherosclerosis and may thus be an indication of intracranial arteries atherosclerosis rather than intracranial arteries stiffness [104]. Indeed, we have no indications on the impact of calcification on intracranial arteries stiffness.
Thus it is difficult to conclude regarding the impact of peripheral arteries stiffness and structure and function of cerebral arterioles. We might expect that an increase in aortic stiffness would lead to similar cerebral arteriolar alterations regardless of the conditions associated with this increase (i.e., aging, hypertension or chronic kidney disease). However, it is worth noting that in these three conditions, cerebral arteriolar dilatation may be impaired following either a decrease in cerebral arteriolar distensibility in aging, remodelling leading to a decrease in internal diameter in hypertension or endothelial dysfunction in chronic kidney disease. This may lead to cerebral hypoperfusion by increasing the lower limit of CBF autoregulation.
3.3 Pulse Pressure and Endothelial Dysfunction
Peripheral arteries stiffness by increasing pulse pressure may impact on cerebral arteriolar endothelial function. As previously mentioned, endothelium-dependent relaxation is impaired in chronic kidney disease [102]. However, there is no indication that this is related to an increase in aortic stiffness. It has already been reported that pulse pressure may impair endothelial function in hypertensive rats [105]. We should acknowledge that the endothelium is under a pulsatile mechanical stress. Some studies have tested the impact of such a stress on endothelial function in vitro but without targeting the cerebrovascular endothelium. Nonetheless, these studies show that a cyclic mechanical stretch of 12 % (1 Hz) imposed to human umbilical vein endothelial cells (HUVEC) grown on a flexible membrane led to the increase in reactive oxygen species (ROS) at 30 min followed by a rise in PKC activity and ICAM-1 expression after 24 h [106, 107]. Activation of NADPH oxidase (p22phox subunit) was shown to be responsible for this increased inflammatory response including a NFκB-dependent TNFα production in response to a 6 % stretch at 1 Hz for 6 h [108] as well as the rise in MMP-2 and -14 activities [109], which are considered in vivo to be necessary for arterial wall remodelling. In addition, cyclic stretch induces a sequential activation of PKC isoform (α and ε) that contribute to Raf/ERK1/2 activation, and PKC-ε appears to play a key role in endothelial adaptation to hemodynamic environment [107]. These experimental data, although only collected in cultured endothelial cells, support in vivo integrative data demonstrating a direct relationship between cerebral artery compliance and endothelial function [110]. At least one study reports a correlation between conduit vessel stiffness and endothelial function. However, the study does not allow to conclude if the increase in conduit vessel stiffness is a cause or a consequence of endothelial dysfunction [111].
3.4 Endothelial Dysfunction and CBF Maladaptation
The role of endothelial dysfunction in chronic and/or acute CBF maladaptation is pre-supposed but difficult to experimentally isolate considering the multiple impact of the function of the endothelium on arterial diameter, wall structure, permeability, and transport of molecules as well as migration of blood cells through the wall [38, 112, 113]. Aging may offer some opportunities to evaluate impact of endothelial dysfunction on CBF maladaptation.
Firstly, the gradual decline of the endothelial cell function characterizes cerebral vascular aging [38, 114, 115]. Infusion of l-arginine (eNOS substrate) increases less blood flow in the MCA of older compared to younger human subjects [116] suggesting impaired eNOS enzymatic activity. Circulating levels of asymmetric dimethylarginine (ADMA), the endogenous eNOS inhibitor, increase with age in humans and could be an independent risk factor of ischemic stroke [117–119]. In Alzheimer’s disease patients, plasma levels of ADMA increase while those of NO decrease [120]. ADMA induce endothelial dysfunction of cerebral arteries [121] and decrease CBF in healthy humans [122]. Yet, one study reported that ADMA concentration in CSF decreases with age [123]. In addition, a rapid inactivation of NO by ROS could attenuate cerebral endothelial function [124–126]. Secondly, aging is associated with a gradual stiffening of the carotid arteries [127], transmitting the pulsations of peripheral flow to the brain microcirculation that favors pulse wave encephalopathy [20]. Thirdly and most importantly, preclinical studies suggest that endothelial dysfunction and alteration in the biomechanical properties of the vascular wall synergize in cerebral arteries [110] (Fig. 7.2). Hence, carotid stenosis [128] and endothelial dysfunction [128–130] may perturb cerebral artery hemodynamics and contribute to the decrease in cerebral capillary density [89], a phenomena that is already detectable during the fifth decade in healthy humans [131]. Because CBF is tightly coupled to neuronal metabolic demand [132–135], the development of chronic brain hypoperfusion together with the increase in intercapillary distances (for review, [136]) promotes progressive hypometabolism [137], chronic BBB inflammation, and leakiness [138], at a stage where brain cell dysfunction is irreversible. Age-related cognitive decline may therefore be strongly influenced by cerebrovascular dysfunctions, a defect that may be magnified by the structural changes of the cerebral artery walls (for review [129, 134, 139–141]).
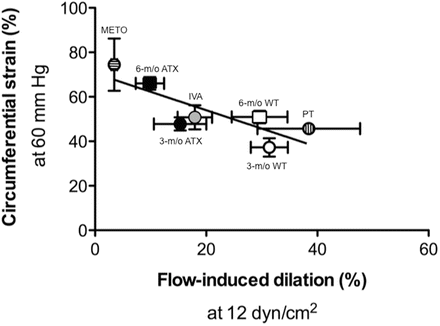
Fig. 7.2
Correlation between endothelial flow-mediated dilation (%) observed at 12.0 ± 0.5 dyn/cm2 and the circumferential strain (%) observed at an intraluminal pressure of 60 mmHg, in cerebral arteries isolated from 3 to 6 months WT mice and severely dyslipidemic (ATX; LDLR−/−; hApoB+/+) mice. Mice were treated for 3 months with either ivabradine (IVA; a I f channel current inhibitor) or metoprolol (METO; a β-adrenergic receptor antagonist), or mice exposed for 3 months to voluntary physical training (PT). There is a negative and significant correlation between both parameters (P < 0.0227; r = 0.824; n = 7), suggesting an interaction between endothelial function and wall structure. Reproduced with permission from [110]
3.5 CBF Maladaptation and Disturbed Brain Metabolism
We pose that defects (functional, mechanical, and structural) in blood flow to the brain leads to a disturbed brain metabolism that would generate neuroglial energy crisis and neuronal loss, together with the accumulation of cytotoxic by-products of brain activity [139, 140, 142]. Consequently, chronic suboptimal brain perfusion would impair cognition [129, 139]. In support of this contention, resting CBF gradually declines in most areas of the brain between the age of 21 and 81 years in healthy volunteers [132]. In addition, oxygen extraction fraction increases in the lower perfused area only as previously shown by others [143, 144]. These brain areas are vulnerable to neuronal degeneration [145] and increased oxygen extraction fraction is a sign of poor oxygen delivery to neurons, and chronically, contribute to the transition from a healthy to an unhealthy brain [132].
4 Challenges and Conclusions
Stiffness impacts on CBF and brain function. The importance of intracranial arteries stiffness remains to be evaluated. Aging, hypertension, and chronic kidney disease are three conditions associated with an increase in cognitive impairment and risk of stroke. However, experimental data suggest that only aging is associated with an increase in cerebral arterioles stiffness. In hypertension, stiffness of intracranial arteries depends on vessels size whereas, at least in an experimental model, chronic kidney disease was not associated with increase or decrease in cerebral arterioles distensibility. In contrast, peripheral artery stiffness is more and more considered as one of the main factor associated with the increased risk of stroke and cognitive impairment during physiopathological conditions such as aging, hypertension, and chronic kidney disease, factor recognized as the pulse wave encephalopathy. Hemodynamic changes such as increase in pulsatility may have an impact on the wall plasticity, on the endothelial function, or on other unidentified mechanism and contribute to brain damages. In consequences, and as emphasized by many authors cited in this chapter, treatment that improves peripheral artery stiffness may be of interest to decrease or prevent the risk of stroke and cognitive impairment rather than treatments that “just” decrease systemic blood pressure.
Acknowledgments
Funding: This research on cerebrovascular aging is supported by the Canadian Institutes of Health Research (MOP 89733), the Montreal Heart Institute Foundation, and the Heart and Stroke Foundation of Quebec.
Conflict of Interest: none.
References
1.
Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844–66. doi:10.1016/j.neuron.2013.10.008.PubMed
2.
Pase MP, Herbert A, Grima NA, Pipingas A, O’Rourke MF. Arterial stiffness as a cause of cognitive decline and dementia: a systematic review and meta-analysis. Intern Med J. 2012;42(7):808–15. doi:10.1111/j.1445-5994.2011.02645.x.PubMed
3.
Pase MP, Pipingas A, Kras M, Nolidin K, Gibbs AL, Wesnes KA, Scholey AB, Stough C. Healthy middle-aged individuals are vulnerable to cognitive deficits as a result of increased arterial stiffness. J Hypertens. 2010;28(8):1724–9. doi:10.1097/HJH.0b013e32833b1ee7.PubMed
4.
Rabkin SW. Arterial stiffness: detection and consequences in cognitive impairment and dementia of the elderly. J Alzheimers Dis. 2012;32(3):541–9. doi:10.3233/JAD-2012-120757.PubMed
5.
Scuteri A, Tesauro M, Appolloni S, Preziosi F, Brancati AM, Volpe M. Arterial stiffness as an independent predictor of longitudinal changes in cognitive function in the older individual. J Hypertens. 2007;25(5):1035–40. doi:10.1097/HJH.0b013e3280895b55.PubMed
6.
Triantafyllidi H, Arvaniti C, Lekakis J, Ikonomidis I, Siafakas N, Tzortzis S, Trivilou P, Zerva L, Stamboulis E, Kremastinos DT. Cognitive impairment is related to increased arterial stiffness and microvascular damage in patients with never-treated essential hypertension. Am J Hypertens. 2009;22(5):525–30. doi:10.1038/ajh.2009.35.PubMed
7.
Waldstein SR, Rice SC, Thayer JF, Najjar SS, Scuteri A, Zonderman AB. Pulse pressure and pulse wave velocity are related to cognitive decline in the Baltimore Longitudinal Study of Aging. Hypertension. 2008;51(1):99–104. doi:10.1161/HYPERTENSIONAHA.107.093674. HYPERTENSIONAHA.107.093674 [pii].PubMed
8.
Watson NL, Sutton-Tyrrell K, Rosano C, Boudreau RM, Hardy SE, Simonsick EM, Najjar SS, Launer LJ, Yaffe K, Atkinson HH, Satterfield S, Newman AB. Arterial stiffness and cognitive decline in well-functioning older adults. J Gerontol Ser A, Biol Sci Med Sci. 2011;66(12):1336–42. doi:10.1093/gerona/glr119.
9.
Lakatta EG. The reality of aging viewed from the arterial wall. Artery Res. 2013;7(2):73–80. doi:10.1016/j.artres.2013.01.003.PubMedPubMedCentral
10.
Wang M, Jiang L, Monticone RE, Lakatta EG. Proinflammation: the key to arterial aging. Trends Endocrinol Metab. 2014;25(2):72–9. doi:10.1016/j.tem.2013.10.002.PubMedPubMedCentral
12.
Katzmarzyk PT, Janssen I. The economic costs associated with physical inactivity and obesity in Canada: an update. Can J Appl Physiol. 2004;29(1):90–115.PubMed
13.
Larson EB, Wang L, Bowen JD, McCormick WC, Teri L, Crane P, Kukull W. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med. 2006;144(2):73–81.PubMed
14.
Lautenschlager NT, Almeida OP. Physical activity and cognition in old age. Curr Opin Psychiatry. 2006;19(2):190–3. doi:10.1097/01.yco.0000214347.38787.37.PubMed
15.
Grundy SM, Hansen B, Smith Jr SC, Cleeman JI, Kahn RA. Clinical management of metabolic syndrome: report of the American Heart Association/National Heart, Lung, and Blood Institute/American Diabetes Association conference on scientific issues related to management. Circulation. 2004;109(4):551–6. doi:10.1161/01.CIR.0000112379.88385.67.PubMed
16.
Koren-Morag N, Goldbourt U, Tanne D. Relation between the metabolic syndrome and ischemic stroke or transient ischemic attack: a prospective cohort study in patients with atherosclerotic cardiovascular disease. Stroke. 2005;36(7):1366–71. doi:10.1161/01.STR.0000169945.75911.33.PubMed
17.
O’Donnell MJ, Xavier D, Liu L, Zhang H, Chin SL, Rao-Melacini P, Rangarajan S, Islam S, Pais P, McQueen MJ, Mondo C, Damasceno A, Lopez-Jaramillo P, Hankey GJ, Dans AL, Yusoff K, Truelsen T, Diener HC, Sacco RL, Ryglewicz D, Czlonkowska A, Weimar C, Wang X, Yusuf S. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case–control study. Lancet. 2010;376(9735):112–23. doi:10.1016/S0140-6736(10)60834-3.PubMed
18.
Solfrizzi V, Scafato E, Capurso C, D’Introno A, Colacicco AM, Frisardi V, Vendemiale G, Baldereschi M, Crepaldi G, Di Carlo A, Galluzzo L, Gandin C, Inzitari D, Maggi S, Capurso A, Panza F. Metabolic syndrome and the risk of vascular dementia: the Italian Longitudinal Study on Ageing. J Neurol Neurosurg Psychiatr. 2010;81(4):433–40. doi:10.1136/jnnp.2009.181743.PubMed
19.
Scuteri A, Cunha PG, Rosei EA, Badariere J, Bekaert S, Cockcroft JR, Cotter J, Cucca F, De Buyzere ML, De Meyer T, Ferrucci L, Franco O, Gale N, Gillebert TC, Langlois M, Laucevicius A, Laurent S, Mattace Raso FU, Morrell CH, Muiesan ML, Munnery MM, Navickas R, Oliveira P, Orru M, Pilia MG, Rietzschel ER, Ryliskyte L, Salvetti M, Schlessinger D, Sousa N, Stefanadis C, Strait J, Van Daele C, Villa I, Vlachopoulos C, Witteman J, Xaplanteris P, Nilsson P, Lakatta EG, Hofman A. Arterial stiffness and influences of the metabolic syndrome: a cross-countries study. Atherosclerosis. 2014;233(2):654–60. doi:10.1016/j.atherosclerosis.2014.01.041.PubMedPubMedCentral
Related posts:
 Hypertension and Brain Inflammation: Role of RAS-Induced Glial Activation
Hypertension and Brain Inflammation: Role of RAS-Induced Glial Activation
 A Systematic Review of Reviews to Examine the Association Between Hypertension and Cognitive Impairments in Adults
A Systematic Review of Reviews to Examine the Association Between Hypertension and Cognitive Impairments in Adults
 Endothelium, the Blood–Brain Barrier, and Hypertension
Endothelium, the Blood–Brain Barrier, and Hypertension
 Cognitive Dysfunction and Dementia in Animal Models of Hypertension
Cognitive Dysfunction and Dementia in Animal Models of Hypertension
 Hypertension and Stroke
Hypertension and Stroke
 Neuroimaging as a Research Tool in Human Essential Hypertension
Neuroimaging as a Research Tool in Human Essential Hypertension
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


