Assessing Risk: Gene Discovery
Thomas Fernandez
Matthew W. State
Introduction
There is indisputable evidence for the heritability of most early-onset psychiatric illnesses. However, the specific genes conveying risk for these disorders remain largely unknown. Given the strength of the data regarding genetic contributions to childhood disorders, the process of gene discovery has been surprisingly slow and frustrating. This, of course, has not been for lack of effort, ingenuity, or determination on the part of patients, families, and researchers. The obstacles that have impeded progress in child psychiatry have presented similar formidable challenges to other medical disciplines that contend with common, multifactorial illnesses. Fortunately, as in these other cases, the fruits of decades of labor in gene hunting are now beginning to be harvested.
Converging forces have resulted in widespread and well justified optimism about the prospects for major discoveries in the genetics of developmental neuropsychiatric disorders: the combination of hard-won insights into the genetic mechanisms underlying neuropsychiatric disorders of childhood, revolutionary advances in genomic technologies, an extraordinary level of involvement among parent and advocacy groups, and broad interdisciplinary research efforts involving clinicians, geneticists, and neuroscientists is transforming the field. Indeed, over the past several years, the first strong evidence for specific genetic contributions to child psychiatric and developmental disorders has been identified. Over the next decade, numerous genetic risks for childhood disorders will be identified, dramatically altering our understanding of the basic mechanisms of child psychiatric disease and fundamentally influencing clinical practice.
In this chapter we will address the challenges to gene identification that have confounded investigations of child psychiatric disorders and outline the major research strategies aimed at overcoming these obstacles. This discussion is intended to lay the groundwork for subsequent chapters, which will review genetic findings relevant to specific disorders, describe the interplay of environment and genetics in conferring both risk and resilience, and highlight the potential contribution of genetics to research in the areas of psychopharmacology and neuroimaging.
Obstacles to Gene Discovery in Childhood Disorders
Multiple Interacting Genes
Over the last three decades, the identification of disease-causing genes has become commonplace (1). These successes have
largely been in the area of single gene disorders exhibiting Mendelian patterns of inheritance: that is, dominant, recessive, or X-linked (Table 2.3.2.1). As a general proposition, the inheritance of psychiatric disorders, such as schizophrenia, bipolar disorder, autism, and Tourette syndrome do not appear to fall into this category, in the sense that they cannot be accounted for by the transmission of a single gene. Rather, the commonly accepted view is that, for the most part, these disorders will be found to be the result of multiple interacting alleles, each with relatively small contributions, compared to the genetic effects observed in Mendelian disorders. For instance, in the case of autism spectrum disorders, analysis of linkage data and family studies has led to widespread acceptance of the hypothesis that at least 15 genes are likely to be involved (2).
largely been in the area of single gene disorders exhibiting Mendelian patterns of inheritance: that is, dominant, recessive, or X-linked (Table 2.3.2.1). As a general proposition, the inheritance of psychiatric disorders, such as schizophrenia, bipolar disorder, autism, and Tourette syndrome do not appear to fall into this category, in the sense that they cannot be accounted for by the transmission of a single gene. Rather, the commonly accepted view is that, for the most part, these disorders will be found to be the result of multiple interacting alleles, each with relatively small contributions, compared to the genetic effects observed in Mendelian disorders. For instance, in the case of autism spectrum disorders, analysis of linkage data and family studies has led to widespread acceptance of the hypothesis that at least 15 genes are likely to be involved (2).
Many psychiatric conditions are also strongly influenced by nongenetic factors. For instance, in the case of schizophrenia, the monozygotic concordance rate (see Table 2.3.2.1) is approximately 50 percent (3,4). The observation that twins sharing all their genetic material share a diagnosis only half the time strongly suggests that influences apart from the sequence of their DNA is contributing to disease risk. These influences may be environmental in the classic sense, or may involve heritable genetic mechanisms that are not coded for in the sequence of DNA (so-called epigenetic factors). The elaboration of the extent and nature of gene–environment interactions in developmental neuropsychiatric disorders is a vibrant area of research that has resulted in important recent insights into the etiopathology of mood and attentional disorders, among others. These findings are reviewed in more detail subsequently in the text.
An Uncertain Genetic Architecture
The combination of multigene inheritance, environmental, and epigenetic influences present significant challenges to researchers interested in gene identification. In addition, fundamental questions remain regarding the genetic architecture of nearly every childhood neuropsychiatric disorder. For instance, it is not known whether genetic variation that is common or rare in the population is likely to carry the lion’s share of risk for common childhood psychiatric conditions. As will be discussed subsequently, this issue is critically important in selecting appropriate research strategies to identify contributing genes as well as in interpreting the resulting findings.
As a general proposition, two alternatives are most commonly investigated: One is the so-called “common disease-common variant” hypothesis, which holds that most of the risk for complex neuropsychiatric disorders will be accounted for by variations, or alleles, that are common in the population. By definition, these are genetic polymorphisms that are present in more than 1 percent of individuals. An often-cited example is Alzheimer’s disease and the increased risk conferred by the e4 allele at the apoliporotein E (APOE) gene locus, a variance that occurs with relatively high frequency in the general population (5,6).
It is widely accepted that in the case of common disorders that begin late in life, common alleles are likely to play a major role. This is because natural selection is likely to favor variations whose effects occur after the age at which individuals typically reproduce. Hence, deleterious alleles contributing to late-onset conditions may attain a substantial frequency in the general population.
This same logic, with minor modifications, can be applied to early-onset disorders: if one presumes that such conditions result from the conspiracy of multiple genes and that each contributing genetic variation has a relatively small effect, selection against any individual allele may be weak, allowing disease-related variations to become common in the population (7,8,9). In addition, it is possible that certain ancient alleles may have historically conferred some selective advantage, and only more recently contributed to disease. This is thought to be the case with respect to the recent identification of a common genetic variant associated with both childhood and adult-onset obesity (10). One can imagine that a gene allele predisposing to higher body mass index could have previously been advantageous during times of scarcity, and that relatively recent environmental changes would have transformed this into a genetic risk.
The common disease-common variant hypothesis is also consistent with what is known about the overall structure of the normal human genome (11). The majority of overall variation within a population is accounted for by common alleles. It would follow then that if selective pressure were not acting to limit the frequency of incrementally contributing loci, common diseases would reflect this underlying structure, i.e., common variation would account for common disease.
An alternate possibility is that a significant proportion of early-onset child psychiatric disorders may be the result of rare genetic variation. This could occur via several scenarios: either through the accumulation of many different rare mutations within one or a small number of genes (so-called allelic heterogeneity), or rare mutations in any of a large number of genes resulting in a similar or overlapping phenotype (locus heterogeneity). Moreover, these two mechanisms, locus and allelic heterogeneity, could combine within what is now considered a single psychiatric syndrome, a scenario which would present significant challenges to progress in genetic research.
The rare variant hypothesis is intuitively attractive, particularly in the case of severe early-onset illnesses. One could imagine that a genetic variation contributing to fundamental impairments in social functioning arising early in life could be subject to selective pressures, as affected individuals might be less likely to have offspring than those who are unaffected.
If one looks to other areas of medicine for clues, it is most likely that a combination of rare and common variants will be found to play a role in child psychiatric illness. For instance, in the case of diseases such as hypertension and breast cancer, rare alleles carrying significant disease risks have been identified. However, these mutations do not appear to account for the majority of population risk. Nonetheless, the discovery of rare alleles has provided vitally important insights into the pathophysiology of these disorders (12,13).
As noted, the question of whether child psychiatric disorders involve common or rare genetic variation is highly relevant because those methods and study designs that may be most appropriate to identify the contribution of one type of risk may have little ability to identify the other. This issue will be addressed as the various approaches to gene hunting are presented below.
Phenotypic Heterogeneity
In addition to the obstacles presented by complex inheritance and an uncertain mode of inheritance, diagnostic issues in child psychiatric disorders present major challenges to geneticists. Ultimately, irrespective of the specific methodology employed, disease-gene hunting involves identifying observable clinical phenomena that bring together individuals with some degree of shared genetic risk and then uncovering the responsible or contributing variation(s) within the genome. If one has difficulty identifying a group of affected individuals who share a proportion of their risk in common, gene identification can be quite difficult.
The absence of reliable and specific physiological markers for childhood psychiatric illnesses presents a significant challenge. Of course, one problem is that our current diagnostic
approaches may have weak correlations with underlying biological mechanisms. It is not yet possible to determine, for instance, whether similar genetic liabilities may underlie several diagnostic categories that are considered as quite separate within the boundaries imposed by current systems of classification (14). The high frequency of comorbidities and wide-ranging clinical presentations seen in child psychiatric syndromes certainly suggests that this may be the case. In addition, while severe forms of any disorder may be quite easily recognized, more subtle manifestations may be difficult to assess and verify. The unambiguous delineation of affected versus unaffected individuals, even within an extended family, can also be complicated by clinical phenomena that change through the course of development, either through an age-dependent onset, a waxing and waning course, or symptoms that decrease markedly in adulthood.
approaches may have weak correlations with underlying biological mechanisms. It is not yet possible to determine, for instance, whether similar genetic liabilities may underlie several diagnostic categories that are considered as quite separate within the boundaries imposed by current systems of classification (14). The high frequency of comorbidities and wide-ranging clinical presentations seen in child psychiatric syndromes certainly suggests that this may be the case. In addition, while severe forms of any disorder may be quite easily recognized, more subtle manifestations may be difficult to assess and verify. The unambiguous delineation of affected versus unaffected individuals, even within an extended family, can also be complicated by clinical phenomena that change through the course of development, either through an age-dependent onset, a waxing and waning course, or symptoms that decrease markedly in adulthood.
TABLE 2.3.2.1 GLOSSARY OF SELECTED TERMS. (COURTESY OF NATIONAL HUMAN GENOME RESEARCH INSTITUTE) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The issue of diagnostic uncertainty in child psychiatry can also pose significant logistical problems in that it can be quite difficult to ascertain comparable samples across sites that are geographically remote. For example, in the case of hypertension, large-scale studies of individuals may be undertaken in which the diagnostic measures may involve little more than multiple readings from a blood pressure cuff. Contrast this with what is typically required for a state-of-the-art psychiatric diagnosis in almost any disorder. One can imagine that the effort and expense required to collect useful data on large numbers of patients in child psychiatry is considerably more challenging than in many other fields of medicine.
Alternatives to categorical approaches to phenotypic assignment aimed at addressing these challenges are discussed in some detail below.
Approaches to Gene Discovery and Characterization
Assessing Heritability and Patterns of Transmission
Most often, gene discovery efforts are preceded by epidemiology investigations aimed at determining the general nature and extent of the genetic risk. Such studies typically seek to identify whether a particular disorder aggregates within families and, if so, whether there is an identifiable pattern of transmission. Such investigations may, for example, evaluate the risk to a relative of an affected individual as compared to the overall incidence of a disorder. The resulting quantity is referred to as λ with a subscript denoting the degree of relatedness. For example a λsib would refer to the relative risk to a sibling of an affected individual. This calculation can be quite useful, first in identifying whether genes might be involved. Moreover, a comparison of this risk among different degrees of relatedness may provide a clue as to the nature of the genetic transmission (15,16).
However, such investigations typically cannot determine whether the observed familial aggregation or increased risk is the direct result of genetic influences. In this regard, both twin and adoption studies play a critical role in teasing apart the relative contribution of genetic factors versus environmental factors in disease etiology. For twin studies, the rates at which monozygotic twins (MZ) share a diagnosis are compared to the rates for dizygotic twins (DZ). An assumption is made that both types of same-sex twins will have a similar degree of shared environmental influences. Consequently, if genes “trump” environment in the etiology of a disorder, those siblings that share all of their DNA (MZ) should be more likely to share a diagnosis than twins who share the same amount of DNA as any sibling pair (DZ). Conversely, if environment predominates as a contributing factor, rates of concordance should not fundamentally differ based on the amount of shared genetic material. Adoption studies accomplish a similar goal by comparing monozygotic twins who are raised together versus those in which twins are “adopted away.” These types of investigations are quite powerful, but are less common in the literature than twin studies.
For autism spectrum disorders (ASDs), MZ concordance has been found to be in the neighborhood of 60 percent for the full syndrome and 90 percent for the broad spectrum. In contrast, DZ concordance has been found to be relatively low, about 3–15 percent, depending on the diagnostic criteria employed. These data support the conclusion that the observed familial clustering is largely the result of genetic factors and translate into an estimate of heritability that places ASDs among the most strongly genetic of all neuropsychiatric conditions (for reviews, see (17,18)).
Once family, twin, and adoption studies have demonstrated that genetic factors are likely to play a role in the pathogenesis of a disorder, there are several different means to identify the specific genes involved. Direct approaches include three overlapping methodologies: linkage analysis, including whole genome screens in affected sibling pairs; gene association studies, including candidate gene studies and family-based studies of association; and cytogenetic methods, including karyotyping, fluorescence in situ hybridization (FISH), and microarray comparative genomic hybridization (CGH). We will next examine the basic principles behind these methods, as well as advantages and pitfalls of each as they relate to the search for disease susceptibility genes in child psychiatry.
Linkage Analysis
Linkage analysis assesses the probability that a given phenotype and a particular genetic marker (or series of markers) are transmitted together from one generation to another (Figure 2.3.2.1 A). To appreciate how the investigation of evenly spaced “anonymous” markers, which are typically known bits of “nonfunctional” DNA, may lead to the identification of a disease gene, one needs to refer to basic genetic principles. When egg and sperm are formed, homologous chromosomes from each of the 22 pairs of autosomes have a tendency to exchange information through a process known as crossing over. Thus, each of the chromosomes in the haploid gamete is, on average, a mixture of its two parental chromosomes. During this process of gamete formation, the likelihood that any two points on a parental chromosome will have a crossover between them is a function of how far apart they are: Loci at opposite ends of a chromosome will be likely to be separated by a crossover. Loci close to each other will be less likely to have a crossover between them and will tend to pass together through multiple generations.
Thus, if one were to investigate a simple dominant disorder due to a gene of major effect using random markers spaced throughout the genome, the closer a particular marker was to the gene mutation causing the disorder, the more likely it would be that that marker would reliably appear in each affected individual (and would not appear in unaffected individuals). Through the investigation of many markers in many affected persons, one can begin to close in on a region of a chromosome containing a disease-related gene. This process is known as positional cloning, given that one is initially narrowing in on a location within the genome without knowing in advance anything about the genes present in that chromosomal segment.
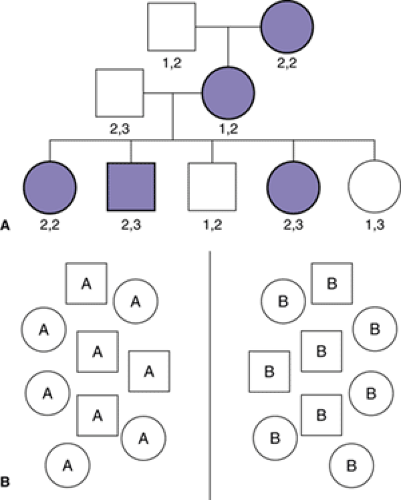 FIGURE 2.3.2.1. Schematic illustrations of linkage and association principles. A. Linkage analysis. A pedigree showing disease (solid shade) among males (squares) and females (circles) in multiple generations. The pair of numbers under each member of the pedigree represents the alleles of a DNA marker which was used to genotype the family. By analyzing the pattern of affected status within pedigrees, one may hypothesize the pattern of disease inheritance (dominant in this case) and then use a parametric linkage approach. By analyzing the frequency with which a particular genotype occurs along with disease in one or multiple families, it may be possible to conclude that a gene involved in disease phenotype is likely to lie in close proximity to a DNA marker. In this case, grandmaternal marker allele 2 appears to be linked to disease. B. Association studies. Individuals with a particular disorder or phenotype (labeled A) are compared with control subjects without the disease or phenotype (labeled B) to determine whether one group is more likely to carry particular allele(s) of the gene(s) being studied. |
As a general matter, two basic approaches to linkage analysis predominate in the hunt for human disease genes. The first is known as parametric linkage analysis. In this case, one specifies a hypothesis about the nature of the proposed genetic transmission (for instance, that a disorder is the result of a dominantly acting mutation that is rare in the population) and then calculates the odds of seeing the observed pattern of transmission given the proposed model versus the odds of observing the same pattern of transmission if there were no linkage (free recombination). Results are expressed as a logarithm of the odds (LOD) score, such that, for example, an LOD score of 3 indicates the odds in favor of linkage between a marker and the disease are 103:1. Indeed, this particular score is taken as the threshold for statistical significance in an investigation of the whole genome, due to the fact that multiple comparisons are conducted (one simultaneously investigates multiple markers). Roughly, 1000:1 odds in favor of linkage at a marker (LOD score of 3) corresponds to a genome-wide p value of 0.05.
Parametric linkage analysis is a tremendously powerful approach to investigate Mendelian disorders as demonstrated by a host of dramatic discoveries, including the identification of genes for Huntington’s disease, hypertension, mental retardation, and various cancers (1,12,19). These successes are directly attributable to the presence of genes of major effect exhibiting simple transmission in the families under study. In those instances in which this approach to linkage has been used successfully in complex disorders, it is typically the result of the identification of a rare family demonstrating inheritance that is simpler than what is presumed to be the norm.
As has been discussed, common child psychiatric disorders are unlikely to be exclusively inherited in a Mendelian fashion. Moreover, while it is presumed that many genes will conspire in any affected individual, the way in which any gene carries such risks or interacts with other genes is not yet know. As a result, some researchers have come to favor an alternative approach known as nonparametric linkage. These investigations do not require the specification of a hypothesis regarding the mode or character of inheritance. Instead, one seeks to identify any region of the genome that is shared among affected related individuals (or not shared in affected–unaffected relative pairs) more often than would be expected by chance. Presently, the most commonly used strategy in this regard relies on affected sibling pairs. Although generally less powerful than parametric linkage, nonparametric analyses are more robust in that they are not sensitive to misspecifications of the mode inheritance. As is the case with parametric studies, results are often presented as LOD scores and there are widely accepted statistical thresholds: LOD scores of greater than 2.2 but less than 3.6 are denoted “suggestive” linkage, scores greater than or equal to 3.6 but less than 5.4 are considered statistically significant, and LOD scores of 5.4 or greater are thought to be highly significant (20). These thresholds can be translated into a more practical metric: A suggestive LOD score of 2.2 is likely to be found by chance (erroneously) once per each genome wide scan; an LOD score of 3.6 will be identified by chance once per every 20 scans (20).
Related posts:
 Child and Family Policy: A Role for Child Psychiatry and Allied Disciplines
Oppositional Defiant and Conduct Disorders
Child and Family Policy: A Role for Child Psychiatry and Allied Disciplines
Oppositional Defiant and Conduct Disorders
 Interpersonal Psychotherapy
Interpersonal Psychotherapy
 Understanding Research Methods and Statistics: A Primer for Clinicians
Suicidal Behavior in Children and Adolescents: Causes and Management
Integrating Behavioral Services into Pediatric Care Settings: Principles and Models
Understanding Research Methods and Statistics: A Primer for Clinicians
Suicidal Behavior in Children and Adolescents: Causes and Management
Integrating Behavioral Services into Pediatric Care Settings: Principles and Models
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


