Fig. 16.1
Glutamate microdomains may be formed by specialized protein complexes found on plasma membranes in extrasynaptic regions where astrocytic and neuronal membranes are opposed to one another. Glutamate is released into the synapse (red shading) where it may bind and activate glutamate receptors. Plasma membrane glutamate transporters localized to perisynaptic astroglial processes bind and transport glutamate (green shading). Glutamate levels in extrasynaptic regions (yellow shading) may be regulated by glutamate spillover from synapses, release of glutamate from astrocytes, as well as reuptake of glutamate by glutamate transporters
Extrasynaptic Glutamate Receptors
The G-protein linked mGluRs have a central role in regulating synaptic glutamate. mGluRs are expressed perisynaptically, and activation of mGluR2/3 receptors decreases presynaptic glutamate release (Schoepp 2001) . Thus, activation of mGluRs may serve as a brake on glutamate spillover, preserving input specificity by diminishing synaptic glutamate levels (Huang and Bergles 2004) . This mechanism has recently been exploited to develop a highly selective mGluR2 agonist that decreases glutamate release (Patil et al. 2007) . This development suggests a role for the pharmacological modulation of glutamate spillover as a treatment for schizophrenia and other illnesses where psychosis is a central feature .
16.3 The Schizophrenia Syndrome
Schizophrenia is most properly considered to be a syndrome, as its specific causes are unknown, and the diagnosis is based on the presence of a heterogeneous collection of signs and symptoms. There is no blood test, specific genetic marker, or cognitive battery that can predict who will develop this illness. The schizophrenia phenotype is characterized by the presence of specific clinical findings, often divided into positive, negative, and cognitive symptoms (Buchanan et al. 2000) .Positive symptoms include hallucinations, which are often auditory. Patients report that they hear voices that are clearly located outside of their heads, most often engaged in a running commentary on their thoughts and behaviors (Kay 1990; Badcock 2010) . Other common positive symptoms include paranoid delusions and disorders of thought processes (Kay 1990; Badcock 2010) . Much more debilitating are the negative symptoms, which are associated with the diminution of normal social behaviors, and include withdrawal, decreased speech, diminished eye contact, decreased or muted facial expression and vocal inflection, and diminished spontaneous movement (Buchanan et al. 2000; Fleischhacker 2000) . Cognitive impairments in this illness include, but are not limited to, deficits in verbal fluency, executive function, and working memory (Rajji and Mulsant 2008; Szoke et al. 2008; Wobrock et al. 2008; Potkin et al. 2009; Zanello et al. 2009) . Few individuals suffering from schizophrenia have all of these symptoms, but the persistence of several characteristic symptoms, like auditory hallucinations, must be present in order for someone to be diagnosed with this disorder (Buchanan et al. 2000) .
16.4 Prevailing Hypotheses of Schizophrenia
For decades, scientists have focused on the dopamine hypothesis of schizophrenia, which postulates that dysregulated dopaminergic neurotransmission is a key feature of the pathophysiology of the illness. The dopamine hypothesis is based on the observation that antipsychotics block D2 receptors, and antipsychotic affinity for these receptors correlates with the ability to attenuate psychotic symptoms. Although numerous studies point to dopaminergic abnormalities in schizophrenia, dopamine dysfunction cannot completely account for all of the symptoms observed, since neuroleptics typically are effective only for the positive symptoms of the illness while negative symptoms and cognitive deficits are relatively refractory to treatment with typical antipsychotics (Joyce and Meador-Woodruff 1997; Laruelle et al. 1999) . Consequently, alternative hypotheses that may help explain the pathophysiology of schizophrenia have been sought.
While the dopaminergic neurotransmitter system was implicated due to the effects of antipsychotic drugs, this system does of course not act in isolation. Dopamine receptors are found throughout the brain where they modulate excitatory and inhibitory neurotransmission via G-protein signaling pathways. Blockade of dopamine receptors in corticolimbic circuits directly alters release of other neurotransmitters including glutamate and γ-aminobutyric acid (GABA). Not surprisingly, extensive postmortem studies have found changes in glutamatergic and GABAergic systems in this illness (Alda et al. 1996; Lewis et al. 2004; Deep-Soboslay et al. 2011; McCullumsmith and Meador-Woodruff 2011) . However, evidence for involvement of glutamate receptor dysfunction, in particular the NMDA-subtype glutamate receptor, suggests a prominent role for glutamatergic abnormalities. NMDA receptor antagonists (but not GABA receptor antagonists) can induce both the positive and negative symptoms of schizophrenia, including cognitive deficits (Javitt and Zukin 1991; Tamminga 1999) . Moreover, these compounds can exacerbate both positive and negative symptoms in schizophrenia (Lahti and Tamminga 1995) . Chronic administration of phencyclidine (PCP)-like compounds induces a persistent psychotic symptomatology (Tsai and Coyle 2002) and reduces frontal lobe blood flow and glucose utilization, which is remarkably similar to the “hypofrontality” described in schizophrenia (Hertzmann et al. 1990) .
Despite these observations, the complexity of schizophrenia is not readily explained by a static neurochemical model. The onset of schizophrenia is typically in late adolescence or early adulthood (Alda et al. 1996) . The onset of positive and negative symptoms in a previously normally functioning person, coupled with a lifetime of waxing and waning symptoms, accompanied by the possibility of a steady decline in social, occupational, and cognitive functioning, has led to longitudinal neurodevelopmental models that take into account genetic and environmental factors (Marenco and Weinberger 2000) . Data supporting the neurodevelopmental hypothesis includes studies suggesting that schizophrenia is associated with late winter births in urban environments, as well as a number of other prenatal, perinatal, and postnatal events (Marenco and Weinberger 2000; Lewis and Levitt 2002) . Schizophrenia is perhaps best considered a disorder of neuroplasticity (McCullumsmith et al. 2004a). Plasticity refers to the ability of a system to effect reversible, long term changes in response to stimuli. Molecular correlates of learning and memory, including LTP and LTD, likely facilitate neuroplasticity in the brain; these processes are significantly impaired in severe mental illnesses, including schizophrenia (Malenka and Nicoll 1999; McCullumsmith et al. 2004b; Talbot et al. 2009) . Glutamate transmission is a central component of LTP and LTD, and hence has a central role in plasticity.
16.5 Glutamatergic Abnormalities in Schizophrenia
A number of studies have evaluated glutamate neurotransmission is schizophrenia using different approaches. In this section we first discuss data from magnetic resonance spectroscopy (MRS), a technique which is largely focused on measuring glutamate, glutamine, and associated metabolites in specific brain regions of living patients. A strength of these studies is that the data are collected from afflicted individuals relatively close in time to the onset of the illness, while postmortem studies are examining brain tissues from more aged individuals who have had a lifetime of psychiatric illness. Next, we will briefly discuss data from postmortem studies, with a particular focus on extrasynaptic glutamate receptors and astroglial transporters.
Magnetic Resonance Spectroscopy Findings in schizophrenia
The balance of studies using MRS to examine glutamate, glutamine, N-acetylaspartate (NAA), and other metabolic intermediates have yielded mixed results (Bartha et al. 1997; Deicken et al. 1997; Theberge et al. 2002; Hutcheson et al. 2012; Kraguljac et al. 2012b) . While a couple of studies have found changes in glutamate in schizophrenia, one large meta-analysis found decreases in the glutamate metabolite NAA in the basal ganglia and frontal lobe (Kraguljac et al. 2012a, 2012b) . Changes in NAA levels suggest abnormalities of glutamate synthesis and/or cycling in schizophrenia (Clark et al. 2006) . A different meta-analysis found decreased glutamate and decreased glutamine in the medial frontal cortex in schizophrenia, suggesting that glutamate neurotransmission is diminished in this illness (Marsman et al. 2011) . One interesting finding from these studies is the loss of correlation between NAA and glutamate levels in subjects with schizophrenia, compared to disease-free control subjects (Hutcheson et al. 2012; Kraguljac et al. 2012a). Taken together with the meta-analyses, these data suggest a significant abnormality in the glutamate/glutamine cycle in limbic circuits in schizophrenia. One limitation of the MRS approach is that it measures all glutamate, glutamine or NAA without regard for it/them being intra or extracellular. For example, there may be a global increase in glutamine in the anterior cingulate cortex, with a large increase in intracellular pools, and a small decrease in extracellular levels.
Abnormalities of Glutamatergic Enzymes in Schizophrenia
There are several key enzymes involved in the glutamate/glutamine cycle as well as the synthesis or break-down of glutamate. Changes in enzymes levels may impact the amount of glutamate available for release from neurons and glial cells. Several studies have found decreased expression of carboxypeptidase II (binding and activity), glutaminase (mRNA), and glutamaine synthetase (mRNA and protein) in limbic regions in schizophrenia (Burbaeva et al. 1999; Goff and Coyle 2001; Laruelle et al. 2003; Bruneau et al. 2005) . Other studies have found increases in glutaminase (mRNA and activity) (Gluck et al. 2002; Bruneau et al. 2005) . While these data support the hypothesis that glutamate synthesis and cycling may be impaired in schizophrenia, all of these studies were done at the regional level, and thus fail to capture the complexity of glutamate synapses at the cellular or subcellular level. For example, there may be diminished expression of glutamate enzymes in astrocytes, but increased expression in pyramidal neurons. Finally, one of the most interesting findings is a decrease in the dipeptidase glutamate carboxypeptidase II (GCP II; also known as NAALADase) activity in the frontal cortex and hippocampus in schizophrenia. GCPII catabolizes N-acetylaspartyl glutamate (NAAG) to glutamate and NAA (Ghose et al. 2004) . These findings are consistent with the MRS data discussed above which found decreased levels of NAA. NAAG antagonizes NMDA receptors, and increased levels (secondary to diminished GCP II activity) might contribute to NMDA receptor hypofunction. One strength of this study is that the authors measured enzyme activity, and not just transcript or protein levels, a technically demanding approach (Tsai and Coyle 2002) .
Glutamate Receptor Abnormalities in Schizophrenia
The observation that PCP may cause a schizophreniform psychosis in persons without a prior diagnosis of schizophrenia led to investigation of ionotropic glutamate receptor expression in schizophrenia. Initial hypotheses were focused on the idea that a loss or hypofunction of NMDA receptor activity would be reflected by diminished expression of NMDA receptor subunits as well as NMDA receptor binding sites. However, on balance, studies of NMDA receptor expression in the postmortem brain in schizophrenia have no clear or consistent pattern of findings (McCullumsmith et al. 2012). For example, there are over 18 studies of NMDA receptor subunit expression in the frontal cortex alone, and other than some changes in binding site expression, the hypothesis that there is deficient NMDA receptor expression stands largely unproven (McCullumsmith et al. 2012) . Similar to NMDA receptors, AMPA and kainate receptor studies generally do not have a consistent pattern of abnormalities other than perhaps changes in AMPA receptor GluA2 subunit expression in the hippocampus (Tamminga 1999; Meador-Woodruff et al. 2001a; Harrison 2004) . Interestingly, administration of PCP, which blocks the NMDA receptor channel, leads to increased glutamate release, which may lead to spillover of glutamate from the synaptic cleft to extrasynaptic areas, activating extrasynaptic (non-NMDA) glutamate receptors. This mechanism may simulate/reflect the condition in schizophrenia where impaired astrocyte function leads to diminished glutamate buffering and reuptake.
Metabotropic Receptor Expression in Schizophrenia
While there are fewer postmortem studies of mGluRs, compared to ionotropic receptors , the data are no less contradictory. For example, mGluR3 protein expression has been reported as increased, decreased and unchanged in the frontal cortex (Gupta et al. 2005; Corti et al. 2007; Ghose et al. 2009; Shan et al. 2012) . Genetic linkage studies suggest that mGluR5 is involved in schizophrenia, and increased mGluR5 and mGluR1 transcript, and mGluR1 protein expression have been found in prefrontal cortex in this illness (Ohnuma et al. 1998; Devon et al. 2001; Gupta et al. 2005; Volk et al. 2010) .
Abnormalities of Glutamate Transporters in Schizophrenia
Several studies have reported region-level changes in the expression of the glial glutamate transporters EAAT1 and EAAT2 in schizophrenia. EAAT1 protein expression was decreased and EAAT1 glycosylation altered in the dorsolateral prefrontal cortex (DLPFC) (Bauer et al. 2008, 2010) . Decreased EAAT1 and EAAT2 protein was found in the superior temporal gyrus, while only EAAT2 protein was decreased in the hippocampus (Shan et al. 2013) . In contrast to these protein studies, increased levels of EAAT1 mRNA were found in the anterior cingulate cortex and thalamus (Smith et al. 2001; Bauer et al. 2008; Rao et al. 2012) , suggesting a compensatory response to diminished glutamate reuptake capacity. Alterations in EAAT2 mRNA have also been reported in the hippocampus (decreased) and neocortex (increased, decreased and unchanged) in schizophrenia (Ohnuma et al. 1998, 2000; Matute et al. 2005; Lauriat et al. 2006; Bauer et al. 2008; Rao et al. 2012) .
The neuronal transporters have also been studied. We have previously reported increased expression of EAAT3 protein and mRNA in the anterior cingulate cortex, while other studies have measured EAAT3 mRNA expression in the frontal cortex (increased), DLPFC (no change) and striatum (decreased) (McCullumsmith and Meador-Woodruff 2002; Lauriat et al. 2006; Nudmamud-Thanoi et al. 2007; Bauer et al. 2008; Horiuchi et al. 2012; Rao et al. 2012) . No changes in EAAT3 protein levels were detected in the superior temporal gyrus or hippocampus (Shan et al. 2013) . These conflicting data for neuronal glutamate transporters mirror the findings of glutamate receptor subunit expression, and are limited by the likelihood that glutamate transporter expression changes may be cell-specific, and change in different directions in different populations of cells. These findings have contributed to reformulation of the glutamate hypothesis of schizophrenia, with the idea that changes in glutamate receptor and/or transporter expression in schizophrenia is not a problem of too much or too little protein expression, but a problem with protein trafficking or signaling. Localization and activity of astroglial-localized glutamate transporters is mediated, in part, by protein-protein interactions. Expression of some of these EAAT interacting proteins has been assessed in severe mental illness.
EAAT Interacting Proteins in Schizophrenia
Several glutamate transporter interacting molecules have been identified, including G-protein suppressor pathway 1 (GPS1), JWA, ARHGEF11, and KIAA0302 (also called beta III spectrin). These molecules can affect glutamate transport function through trafficking , anchoring, phosphorylation, glycosylation, and degradation of transporters in the brain. For example, GPS1 decreases EAAT2 mediated glutamate reuptake through a direct protein-protein interaction, and levels of GPS1 protein were elevated in the frontal cortex in schizophrenia. These data suggest that there may be normal levels, but decreased activity, of a specific transporter due to modulation of transporter function or localization to the plasma membrane (Bauer et al. 2008).
In summary, changes in glutamate neurotransmission in schizophrenia point towards complex abnormalities of protein localization and function, contributing to the molecular neuropathology that underlies the schizophrenia phenotype.
16.6 Glutamate Spillover in Schizophrenia
Glutamate Spillover in Schizophrenia
As outlined above, several postmortem studies have found changes in EAAT expression in schizophrenia, as well as changes in the molecules that regulate EAAT localization and activity (Ohnuma et al. 1998, 2000; Smith et al. 2001; McCullumsmith and Meador-Woodruff 2002; Lauriat et al. 2006; Bauer et al. 2008) . In general, these changes in gene expression are consistent with diminished regional expression of astroglial (but not neuronal) glutamate transporter expression and activity. Further, a recent genetic study analyzing copy number variants reported a subject with schizophrenia with a deletion of several EAAT1 exons (Cook and Scherer 2008) . Finally, characterization of the complete GLAST (EAAT1) knockout found changes consistent with behavioral endophenotypes associated with schizophrenia, including locomotor hyperactivity and abnormal social behavior (Karlsson et al. 2008, 2009) . Several of these abnormal behavioral findings were reversed by administration of antipsychotic medication or mGluR2/3 receptor agonist administration, which decreases presynaptic glutamate release. These data suggest that there are region specific deficits in EAAT reuptake capacity in schizophrenia which could lead to glutamate spillover.
Chronic Glutamate Spillover May Lead to Remodeling of Synapses
In the prefrontal cortex (PFC) in schizophrenia, there are changes in the structure, composition, and numbers of excitatory synapses (Broadbelt et al. 2002; Lewis et al. 2003, 2008) . Increased packing density, decreased numbers of dendritic spines and diminished expression of structural proteins suggest significant alterations of synapses in this region (Selemon et al. 1995; Rajkowska et al. 1998, 2002) . Several reports have found specific alterations in layers III and IV of the PFC, including abnormalities of pyramidal cells and interneurons (Hashimoto et al. 2003; Dong et al. 2005; Huang and Akbarian 2007; Huang et al. 2007; Lewis et al. 2008) . One well replicated finding is a decrease in parvalbumin positive interneurons in the middle cortical layers (Lewis et al. 2001; Hashimoto et al. 2003) . A lamina specific deficit in inhibitory tone could lead to increased release of glutamate, which combined with diminished reuptake capacity could lead to increased glutamate spillover.
Accumulating evidence from postmortem gene expression studies suggests neurochemical alterations consistent with spillover. We have found increased mGluR2/3 protein in the PFC (Gupta et al. 2005) , which may be interpreted as an attempt by synapses to decrease spillover by decreasing presynaptic release. Expression of the cystine/glutamate antiportercatalytic subunit (xCT) was also increased in the DLPFC (Baker et al. 2008) . This molecule is expressed on glia and releases glutamate into the extrasynaptic space in exchange for uptake of cystine, which is required for glutathione synthesis. The effect of increased xCT protein expression on glutamate release is not known, because it is the activity, and not expression level, of this molecule that determines the rate of glutamate release. However, changes in the expression of xCT minimally suggest abnormalities of the regulation of extracellular glutamate levels (Baker et al. 2008) . Finally, anumber of studies have also described changes in ionotropic glutamate receptor binding site expression, suggesting a change in NMDA and AMPA receptor stoichiometry in the frontal cortex in schizophrenia (Akbarian et al. 1996; Healy et al. 1998; Ibrahim et al. 2000; Meador-Woodruff et al. 2001b; Beneyto and Meador-Woodruff 2006; Beneyto et al. 2007; Harney et al. 2008) . Interestingly, preclinical studies have shown that glutamate spillover is associated with alterations in ionotropic receptor subunit composition and function (Lozovaya et al. 2004b; Harney et al. 2008) .
We propose that there is remodeling of glutamate synapses in schizophrenia secondary to glutamate spillover (Fig. 16.2). Glutamate spillover may be secondary to increased release (in a misguided attempt to activate “sick” NMDA receptors), as well as deficits in glutamate reuptake capacity. In this setting, we would predict that perisynaptic localization of glutamate transporters on the plasma membrane of astrocytes is diminished, either as a primary deficit in transporter localization, as a compensation for increased extrasynaptic glutamate release, or both. Redistribution of glutamate transporters on astrocytes would lead or contribute to increased spillover, causing excitotoxicity and loss of input specificity. Further, we postulate that these deficits are initially relatively subtle, but chronic in nature, leading to inappropriate remodeling of excitatory synapses which do not function normally. This idea is supported by the phenotype of the GLAST/EAAT1 knockout mice, which have moderate cognitive and behavioral impairment, but no morbidity or mortality associated with seizures (Watase et al. 1998; Karlsson et al. 2008, 2009) .
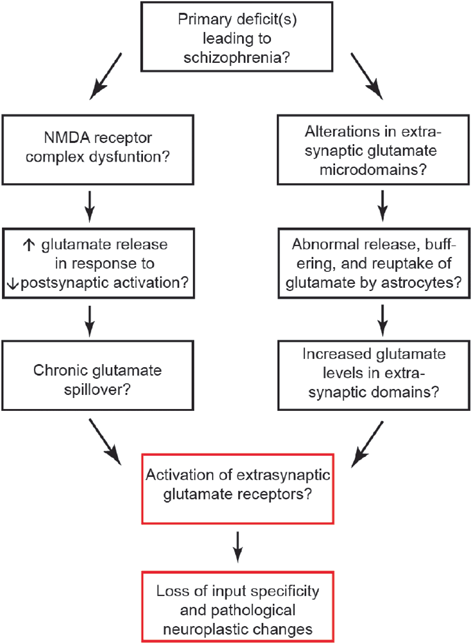
Fig. 16.2
There are likely a number of primary causes which yield the constellations of symptoms found in schizophrenia. Proximate causes might include NMDA receptor dysfunction or abnormalities of glutamate microdomains. The red boxes may be a final common pathway that contributes to the phenotype of this syndrome
16.7 A Role for Glutamate Microdomains
Glutamate Microdomains
Related posts:
 General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
 Ionic Signaling in Physiology and Pathophysiology of Astroglia
Ionic Signaling in Physiology and Pathophysiology of Astroglia
 Enteric Glial Cells: Implications in Gut Pathology
Enteric Glial Cells: Implications in Gut Pathology
 Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
 Novel Therapeutic Approaches to Malignant Gliomas
Novel Therapeutic Approaches to Malignant Gliomas
 Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


