Fig. 7.1
Pyramidal neurons that do not express any functional membrane-bound aquaporins are osmoresistant. In contrast, aquaporin rich astrocytes are clearly osmoresponsive. Two photon laser scanning microscopy (2PLSM) imaging of dendrites, pyramidal neuronal cell bodies and astrocytes (bottom three rows; green, enhanced green fluorescent protein) in cortical brain slices showed that changes in intrinsic optical signals (top row) caused by osmotic challenge were associated with changes in astrocytic volume while neurons were not affected. Normosmotic measurements (column 1, control) were taken prior to 15 min of hypo-osmolar treatment (column 2, − 40 mOsm) that increased light transmittance (LT, ΔT/T %) by neocortical gray matter, denoting tissue swelling. Dendrites and neuronal cell bodies did not change volume, whereas astrocytes swelled. Superfusion of slice with hyperosmotic artificial cerebrospinal fluid (ACSF) (column 3, + 40 mOsm, 15 min) exposed the slice to an 80 mOsm step increase, that greatly reduced LT in the gray matter, signifying tissue shrinkage, but again dendrites and neuronal soma did not shrink, as astrocytes volume clearly decreased. Back in control ACSF (images are not shown), LT levels returned to near baseline as did the astrocytes. Following 10 min of oxygen-glucose deprivation (OGD), the gray matter displayed a propagating wave of elevated LT (not shown) caused by spreading depolarization-induced cell swelling. Pyramidal cell bodies and astrocytes swelled (column, 4). Dendrites also swelled but quickly form beads that reduced LT (Obeidat et al. 2000). Beading is considered an extreme version of focal dendritic swelling that paradoxically lowers LT in swollen tissue because the beads readily scatter light. This overcomes the elevated LT signal caused by neuronal and astroglial swelling. (Modified with permission from Andrew et al. (2007))
The reasons why neurons lacking aquaporins suddenly swell when strongly depolarized as during spreading depolarization represent an interesting question. One possibility is that bulk water influx might occur through pannexin 1 ion channels or other large-pore channels opened by ischemia (Anderson et al. 2005; Thompson et al. 2006) . Pannexin 1 (Px1) is expressed predominantly in neurons where it forms large-pore channels that can pass substances up to 1000 Da (MacVicar and Thompson 2010) . Px1 can be open by depolarization (Pelegrin and Surprenant 2006; Thompson et al. 2008) , high extracellular [K+]o (Silverman et al. 2009) , strong elevation of intracellular [Ca2+]i (Locovei et al. 2006) and by mechanical stretch (Bao et al. 2004) . All of these conditions are present during spreading depolarization. It should be noted, that Px1 activation is not directly responsible for spreading depolarization generation (Madry et al. 2010; Bargiotas et al. 2011) , but rather Px1 functions to maintain depolarization (MacVicar and Thompson 2010; Weilinger et al. 2012) . Indeed, Px1 channels could be directly activated by ischemia in acutely isolated hippocampal neurons (Thompson et al. 2006), by ischemia-induced spreading depolarization in brain slices (Weilinger et al. 2012) and in vivo by spreading depolarization (Karatas et al. 2013) . Px1 activation mediates neuronal death in vitro (Orellana et al. 2011) , but most importantly in vivo (Bargiotas et al. 2011) during focal stroke. Hence, it might be anticipated that Px1 opening should make the neuronal membrane which poorly conducts water at its resting potential instantly permeable to water molecules during spreading depolarization leading to neuronal soma swelling accompanied by focal dendritic swelling (beading) and spine loss.
The synergistic contribution of Px1 channels and cotransporters to the neuronal and dendritic swelling with spine loss is another possibility. Several of the cotransporters that flux significant amounts of water are expressed in the neuronal membrane so cotransporters may also be responsible for water accumulation as well as recovery when water might be quickly translocated from neurons by cotransport proteins (MacAulay and Zeuthen 2010; Zeuthen 2010; Jourdain et al. 2011) . The K+/Cl− contransporter KCC2, the Na+/K+/2Cl− cotransporter NKCC1 and the monocarboxylate transporter MCT2 are the dominant forms localized in neurons (Payne et al. 2003; Pierre and Pellerin 2005; Blaesse et al. 2009) . The amount of water molecules cotransported per the protein turnover is about 500 and notably water can also be transported against an osmotic gradient (MacAulay and Zeuthen 2010) . These cotransporters are bi-directional and can carry a net ion and water influx or efflux, dictated by the free energy transmembrane gradients for the transported ions (Russell 2000; Rocha-Gonzalez et al. 2008; Blaesse et al. 2009) or H+/lactate (Pierre and Pellerin 2005) .
7.6 Neuronal Response to Spreading Depolarization
Since most excitatory synapses in adult brain occur on the dendritic arbor (Harris and Kater 1994) , dendrites were predicted to be the initial site of acute excitotoxic injury leading to neuronal damage and death (Bindokas and Miller 1995) . Indeed, sustained high calcium levels that were shown to develop in distal dendrites upon excitotoxic insult are capable of slowly spreading to the soma, ultimately resulting in acute neuronal injury (Shuttleworth and Connor 2001; Vander Jagt et al. 2008) . Likewise, in the course of spreading depolarization dendritic calcium raises into the micromolar range (Dietz et al. 2008) and prolonged NMDA receptors activation during repolarization appears to underlie these calcium elevations (Aiba and Shuttleworth 2012) . The formation of focal swelling or beading along the dendritic shaft separated from each other by thin dendritic segments (“beads-on-a-string” appearance) is an early morphological sign of acute neuronal injury typically associated with a variety of pathological conditions. Previously, dendritic beading has been reported in the ischemic condition (Hsu and Buzsaki 1993; Hori and Carpenter 1994; Zhang et al. 2005) and without recovery of sufficient blood flow it is a reliable indicator of an irreversible acute terminal injury to fine synaptic circuitry (Zhang and Murphy 2007; Li and Murphy 2008; Risher et al. 2010), and it is an early sign of cell death pathway activation (Enright et al. 2007) .
Spreading depolarization-induced dendritic morphological alterations in the normoxic cortex are equivocal. Indeed, recent in vivo two photon laser scanning microscopy (2PLSM) studies have directly shown that in normal neocortex under physiological conditions, spreading depolarization can be accompanied by a complex pattern of dendritic beading or no beading (Takano et al. 2007; Sword et al. 2013) . Importantly, several rounds of spreading depolarization caused no accumulating dendritic injury in normal neocortex as dendrites fully recovered from beading during repolarization (Sword et al. 2013) . In normal cortex spreading depolarization is the pathophysiological correlate of the migraine aura (Lauritzen 1994) , but even here it can cause a shortage of energy supply (Hashemi et al. 2009) and can be accompanied by a short period of tissue hypoxia with a complex pattern of distribution across the capillary bed (Takano et al. 2007; Yuzawa et al. 2012) . This could possibly translate into a complex pattern of spreading depolarization-induced dendritic beading (Takano et al. 2007; Sword et al. 2013) thus reflecting patterns of tissue hypoxia across the capillary bed with perhaps no beading occurring in areas of luxury oxygen supply in the vicinity of penetrating arterioles (Kasischke et al. 2011) .
Using a photothrombotic model of focal stroke other in vivo 2PLSM studies have shown that without recurrent spreading depolarizations injury to dendritic structure is gated by the degree of ischemia after local blood flow loss (Zhang et al. 2005; Risher et al. 2010) . During severe ischemia (~ 90 % reduction of blood flow) dendritic structure was lost within 10–40 min, but during moderate ischemia (~ 50 % reduction of blood flow) dendritic beading developed within several hours (Zhang et al. 2005) . Dendritic structure could be maintained in the ischemic tissue in the presence of a flowing capillary ~ 80 μm away (Zhang and Murphy 2007), and arterioles can supply oxygen even over longer distances, well above 100 μm (Kasischke et al. 2011) . Spreading depolarization recorded in vivo under conditions of severe metabolic compromise, such as during global ischemia, rapidly damages fine synaptic circuitry (Murphy et al. 2008) and in the absence of reperfusion, as seen in the ischemic core, dendrites remain terminally beaded and spines are lost (Zhang and Murphy 2007; Risher et al. 2010) .
Recurrent spontaneous spreading depolarizations greatly accelerated acute terminal dendritic injury in the moderately ischemic penumbra (Risher et al. 2010) (Fig. 7.2). Dendrites rapidly (< 6 s) beaded and some spines were lost in concert with recurring spreading depolarizations. Dendrites quickly (< 3 min) recovered between spreading depolarizations to near-control morphology, but it is conceivable that signals leading to neuronal death were triggered during this time. Hence, uncoupling of dendritic beading from spreading depolarizations could protect dendrites by delaying terminal injury (Risher et al. 2011) . Preservation of dendrites and integrity of synaptic circuitry is the best indicator of the efficacy of neuroprotection strategy because preservation of neuronal soma alone but not dendrites does not result in proper function (Iyirhiaro et al. 2008) .The likelihood of rapid dendritic recovery between spreading depolarizations was correlated with the presence of nearby flowing blood vessels. However, in about 1/3 of cases, dendrites did not recover even near a flowing capillary, suggesting that the energy demand of widespread repolarization following each spreading depolarization exceeded the energy supply of compromised blood flow. The accumulating stress of repeated spreading depolarizations eventually resulted in terminal dendritic injury (and thus permanent neuronal damage) dramatically accelerating recruitment of the penumbra into the infarct even if residual blood flow was present.
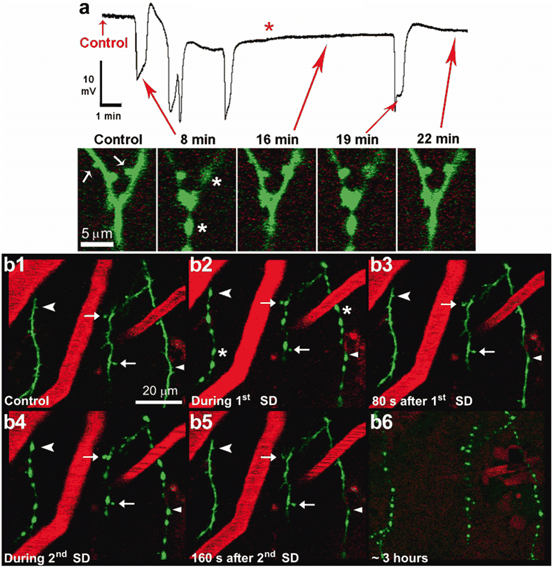
Fig. 7.2
Dendrites undergo a rapid cycle of beading and recovery coinciding with the passage of spreading depolarizations in the ischemic penumbra after focal photothrombosis in mice. a A high-magnification in vivo 2PLSM image sequence of a branched dendrite with red arrows corresponding to various time points on the above recording of spontaneous spreading depolarizationsfrom microelectrode placed next to imaged dendrites. Dendritic spines are indicated by white arrows and asterisk indicates beading (8 min). The dendrite recovers at 16 min, but it is beaded again with spines lost by a subsequent spontaneous spreading depolarization at 19 min. Spontaneous spreading depolarizations are superimposed on a shallow ultraslow negativity (red asterisk) assumed to be an electrophysiological index of the cell death. b Low-magnification image sequence showing dendrites (green, EGFP) and blood vessels (red, labeled with Texas Red dextran) from layer I of the somatosensory cortex. Blood flow within vessels is indicated by streaking caused by scanning of moving non-fluorescent red blood cells. The dendrites appear normal during control (b1) and undergo rapid beading (asterisks) and recovery coinciding with recurrent spreading depolarizations (b2, b4) similarly to the example in (a). Some dendritic spines are permanently lost (arrowhead), transiently lost (chevrons), or persistent (arrows) during spreading depolarization-induced dendritic beading. Appearance of dendritic beads is robust during passage of spreading depolarizations. Finally, nearly 3 h after photothrombosis, the dendrites undergo terminal beadingcoinciding with the passage of spreading depolarization and are no longer able to recover (b6) (vessels are no longer clearly seen at this time point due to Texas Red leaking out, necessitating a reduction in the red channel gain). (Modified with permission from Risher et al. (2010))
As it has been discussed above the most likely mechanism underlying spreading depolarization-induced dendritic beading results from a calcium-independent event such as the excessive influx of ions and obligatory osmotic uptake of water. Certainly, maintained depolarization during intense cooling results in dendritic beading with distinctive ultrastructural characteristics of focally swollen dendrites such as dramatically swollen organelles and electron lucent watery cytoplasm with holes indicating water entry (Kirov et al. 2004) . Currently, there are no quantitative serial section electron microscopy (EM) studies directly examining dendritic disruption arising from spreading depolarization. Only a few single section EM studies have investigated in vivo changes in dendrites immediately after global cerebral ischemia without reperfusion (Yamamoto et al. 1986, 1990; Tomimoto and Yanagihara 1994) . It is possible that ischemia-induced spreading depolarization has invaded neuropil containing examined by EM dendrites, but this is unknown. Regardless, these EM analyses have only provided incomplete information about the extent of acute damage, limited mostly to mitochondrial swelling and disintegration of microtubules indicating calcium entry. Apparently, dendritic microtubules disassemble when intracellular calcium increases to the micromolar range, but although calpain can cleave microtubules, this calcium-activated protease was not involved in initial excitotoxin-induced dendritic beading as shown in brain slices (Hoskison and Shuttleworth 2006) . Yet, the microtubule-stabilizing compound taxol prevented microtubule fragmentation and excitotoxin-induced dendritic beading (Hoskison and Shuttleworth 2006), suggesting that both the initial disruption of microtubules and beading may directly result from excessive water influx. It should be noted, however, that the shortage of metabolic energy associated with spreading depolarization may also result in unregulated polymerization of monomeric G-actin to polymeric F-actin (Atkinson et al. 2004) . Such net conversion of G-actin to F-actin could contribute to dendritic structural rearrangements as beads are known to contain polymerized actin that redisperses during recovery from energy deprivation (Gisselsson et al. 2005) .
7.7 Astroglial Response to Spreading Depolarization
Dendritic spines are present postsynaptically at the majority of excitatory synapses in the brain, where they are often partially ensheathed by astroglial processes to form the tripartite synapse (Harris and Kater 1994; Araque et al. 1999; Ventura and Harris 1999; Witcher et al. 2007, 2010) . Through this close contact, astrocytes provide support for neurons via trophic factors (Yamagata et al. 2002) and maintain healthy synapses by clearing extracellular glutamate, thereby preventing neurotransmitter excitotoxicity (Anderson and Swanson 2000) . Astroglial endfeet wrap around and completely ensheathe blood microvessels (Mathiisen et al. 2010) , providing a critical link between neurons and the blood supply as part of the neuro-vascular unit (Haydon and Carmignoto 2006) . Therefore, in recent real-time in vivo 2PLSM imaging study we have examined spreading depolarization-induced astroglial structural changes concurrently with signs of neuronal injury in the early periods of focal and global ischemia (Risher et al. 2012) . In the penumbra, astrocytes underwent irreversible swelling in conjunction with spreading depolarization despite rapid dendritic recovery (Fig. 7.3). Astroglial swelling was often exacerbated in a stepwise manner by recurrent spontaneous spreading depolarizations and the magnitude of swelling strongly correlated with the total duration of depolarization . Such spreading depolarization-induced persistent astroglial swelling would greatly lessen their neuroprotective and supportive capabilities (Kimelberg 2005) , making neurons more susceptible to damage from the propagating depolarization. Indeed, selective impairment of oxidative metabolism in astrocytes by inhibitors of the mitochondrial enzyme aconitase induces rounds of spreading depolarization sufficient to cause neuronal death (Largo et al. 1996) . Astrocytes are relatively resistant to hypoxia but become quickly injured if hypoxia is combined with acidosis and increased extracellular [K+]o (Chesler 2005) . It is conceivable that spreading depolarization-induced acidosis and elevated extracellular [K+]o combine with hypoxia to compromise astroglial viability, promoting damage to neighboring neurons and thus contributing to a stepwise expansion of the infarct (Nedergaard and Dirnagl 2005) .
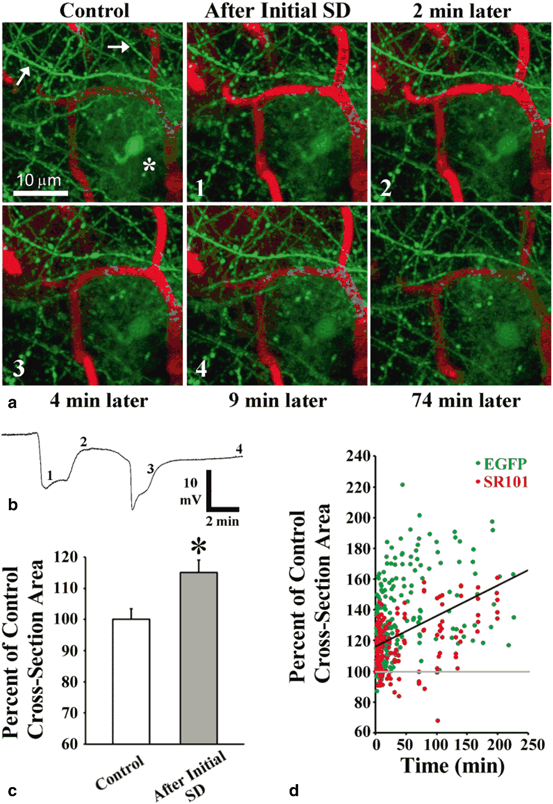
Fig. 7.3
Long lasting spreading depolarization-induced astroglial swelling in the ischemic penumbra after focal photothrombotic stroke. a 2PLSM in vivo images of an astrocyte (asterisk) along with nearby dendrites (arrows) and blood vessels (red, labeled with Texas Red Dextran) in layer I of mouse somatosensory cortex. Each numbered image corresponds with a time point indicated on the respective potential recording at the imaging site shown in (b). Negative deflections in the potential represent spreading depolarizations. The dendrites bead during initial photothrombotically-induced spreading depolarization and rapidly recover (< 2 min). By contrast, the astrocyte swells with no recovery seen. A spontaneous spreading depolarization occurring 4 min later is accompanied by another round of dendritic beading and recovery (shown at 9 min), while the astrocyte continues to swell. c Summary from 64 astrocytes in 20 animals showing the increase in astroglial soma size immediately after initial photothrombotically-induced spreading depolarization (posts-spreading depolarization images were taken 2.3 ± 0.2 min after initial depolarization onset). *P < 0.001, paired t-test. d Somata of EGFP-expressing astrocytes (green dots) and astrocytes labeled with sulforhodamine 101 (SR101) (red dots) continued to increase in size after initial spreading depolarization onset. Values are shown as percent of control cross-section soma area for each astrocyte. Regression line is shown (black) (r = 0.43, P < 0.001; 66 astrocytes from 21 animals). (Modified with permission from Risher et al. (2012))
In brain slices under normoxic conditions astrocytes undergo a passive transient swelling (Risher et al. 2009) in response to the spreading depolarization-elicited rise of extracellular [K+]o (Zhou et al. 2010) . Similarly, spreading depolarization results in transient astroglial swelling in vivo in healthy metabolically uncompromised neocortex (Risher et al. 2012). This transient astroglial swelling in normal healthy cortex is likely reflecting the protective work of these cells during the removal of extracellular [K+]o and glutamate (Mongin and Kimelberg 2005) . Yet here in healthy neocortex, astrocytes could be a critical link in the complex cascade of events linking spreading depolarization to activation of the trigeminal pain fibers that triggers migraine headache. This was recently found when spreading depolarization-induced transient opening of neuronal pannexin 1 channels resulted in the release of pro-inflammatory mediators with subsequent activation of nuclear factor κB (NF-κB) in astrocytes (Karatas et al. 2013) . Activation of NF-κB could cause the formation and continuous release of cytokines, prostanoids, and NO to the subarachnoid space by astrocytes forming glia limitans and promote sustained stimulation of trigeminal nerves around pial vessels.
A transient global ischemia-elicited spreading depolarization induced either persistent or transient astroglial swelling alongside dendritic beading that was reversible with immediate reperfusion (Risher et al. 2012). Astroglial swelling was persistent during severe ischemia characterized by a long-lasting depolarization. Interestingly, when astroglial depolarization during oxygen/glucose deprivation in slices was combined with pharmacological blockade of ATP synthesis, astrocytes were not able to recover their membrane potential during re-oxygenation/normoglycemia, indicating irreversible damage (Xie et al. 2008) . Perhaps these slice experiments closely mimic global ischemia when the sudden reduction in ATP and failure of the sodium-potassium pump results in long-lasting spreading depolarization, leading to persistent swelling of severely metabolically compromised astrocytes. In this respect, such an outcome is analogous to previously reported persistent swelling of astrocytes during cardiac arrest and subsequent terminal spreading depolarization (Risher et al. 2009) . Similarly to normoxic tissue, astroglial swelling was transient during short global ischemic periods distinguished by a short-lasting spreading depolarization. Therefore, it appears that the spreading depolarization-induced transient or persistent nature of astroglial swelling likely depends on the metabolic status of the surrounding tissue.
Astrocytes play a critical role in the clearance of potassium from extracellular space (Kofuji and Newman 2004) by several molecular mechanisms including inward rectifying K+ channels, the Na+/K+/2Cl− cotransporter NKCC1 and the sodium-potassium pump (Mazel et al. 2002; Macaulay and Zeuthen 2012) . Spreading depolarization-elicited astroglial swelling reflects the clearance of the accumulated extracellular potassium (Zhou et al. 2010) with associated water influx through aquaporins, mainly aquaporin 4 (Amiry-Moghaddam and Ottersen 2003; Kimelberg 2005; Leis et al. 2005) . Accordingly, transgenic aquaporin 4-deficient mice display reduced edema and improved neurological outcome after experimental stroke (Manley et al. 2000) . Activation of astroglial metabotropic glutamate receptors increases the water permeability of aquaporin 4 (Gunnarson et al. 2008) . Water influx through aquaporin 4 triggers Ca2+ signaling in astrocytes and results in the activation of signaling cascades that could compromise astrocyte viability and exacerbate the pathological outcome (Thrane et al. 2011) . Thus, astroglial swelling can be harmful and decrease the ability of astrocytes to maintain normal homeostatic function (Kimelberg 2005; Rossi et al. 2007) .
Multiple mechanisms may participate in spreading depolarization-induced astroglial swelling. In normoxic brain tissue with a functional sodium potassium pump, intracellular water translocation by glutamate transporters during glutamate reabsorption and by NKCC1 cotransporter during the clearance of extracellular potassium should contribute to astroglial swelling during spreading depolarization (MacAulay and Zeuthen 2010) . An increase in extracellular potassium during onset of spreading depolarization should provide a favorable driving force for the reversed direction of the transport by K+/Cl− contransporters with associated water influx and thus also contribute to the astroglial swelling (Macaulay and Zeuthen 2012). Then during repolarization heavy [Cl−]i load should provide a favorable driving force for net extrusion of K+, Cl− and water by K+/Cl− contransporters aiding in the fast volume recovery.
Under ischemic conditions when the Na+ and K+ gradients run down due to inhibition of the sodium-potassium pump, voltage-sensitive Na+-coupled transporters such as glutamate transporters will slow down, diminishing their contribution to the astroglial swelling during spreading depolarization (MacAulay and Zeuthen 2010). However, there is a strong indication that activity of the NKCC1 cotransporter is maintained even under disrupted ion gradients (Chen et al. 2005) . Spreading depolarization-elicited extracellular acidification and increased lactate should also add to astroglial water accumulation due to the cotransport of water by the astroglial monocarboxylate transporter during lactate/H+ clearance from the extracellular space (MacAulay and Zeuthen 2010) . Intracellular acidification will similarly facilitate astroglial swelling mainly through activation of Na+/H+ and HCO3 −/Cl− exchangers, leading to accumulation of intracellular osmolytes (Mongin and Kimelberg 2005) . Importantly, Gibbs-Donnan forces should play an important role in the redistribution of ions across membranes of metabolically compromised astrocytes, leading to an uptake of the high extracellular K+ followed by osmotically obligated water through aquaporins and thus persistent astroglial swelling (Kimelberg 2005) .
Astroglial swelling may be a precursor to several detrimental consequences of ischemia , since swelling activates one or more membrane permeability pathways and leads to the release of neuroactive and neurotoxic molecules, including the excitatory amino acids glutamate and aspartate (Kimelberg et al. 1990; Abdullaev et al. 2006; Hines and Haydon 2013) , ATP (Mongin and Kimelberg 2002; Liu et al. 2008; Orellana et al. 2011) and D-serine (Schell et al. 1995; Katsuki et al. 2004) . Such release of ATP and glutamate through astroglial connexin 43 hemichannels could mediate neuronal death through activation of neuronal pannexin 1 channels (Orellana et al. 2011) . Furthermore, spreading depolarization-induced astroglial swelling shown at such early time points after stroke may also be a prelude to devastating events occurring later on, as swelling eventually results in the rupture of the plasmalemma and organelles during delayed astroglial necrosis (Gurer et al. 2009) . Loss of astrocytes would then be a crippling blow to the continued survival of neurons, since it has been well-established that neurons are dependent on the support provided by their glial neighbors both during development and following brain injury (Barreto et al. 2011) .
Alternatively, astroglial swelling may be reflective of the protective work of these cells, including the removal of extracellular K+ and glutamate (Mongin and Kimelberg 2005) , increased glycogen metabolism (Hertz et al. 2007; Seidel and Shuttleworth 2011) and the release of glutathione to defend neurons from oxidative injury (Dienel and Hertz 2005) . Equally, the release of ATP (Kimelberg et al. 2006) and subsequent extracellular accumulation of adenosine (Pascual et al. 2005) could provide neuroprotection by delaying the onset of spreading depolarization (Canals et al. 2008) . This dual role of astroglial swelling may underlie the transition between injury and repair in tissue at risk and the ability of astrocytes to protect surrounding neurons may likely depend on the level of metabolic compromise.
Acknowledgments
The author is grateful for the support provided by the NIH (NS083858) and American Heart Association (12GRNT16570006).
References
Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK, Mongin AA (2006) Pharmacological comparison of swelling-activated excitatory amino acid release and Cl- currents in cultured rat astrocytes. J Physiol 572:677–689PubMedPubMedCentral
Agre P, King LS, Yasui M, Guggino WB, Ottersen OP, Fujiyoshi Y, Engel A, Nielsen S (2002) Aquaporin water channels-from atomic structure to clinical medicine. J Physiol 542:3–16PubMedPubMedCentral
Aiba I, Shuttleworth CW (2012) Sustained NMDAR activation by spreading depolarizations can initiate excitotoxic injury in metabolically compromised neurons. J Physiol 590(Pt 22):5877–5893PubMedPubMedCentral
Aitken PG, Tombaugh GC, Turner DA, Somjen GG (1998) Similar propagation of SD and hypoxic SD-like depolarization in rat hippocampus recorded optically and electrically. J Neurophysiol 80:1514–1521PubMed
Ames A 3rd (2000) CNS energy metabolism as related to function. Brain Res Brain Res Rev 34:42–68PubMed
Amiry-Moghaddam M, Ottersen OP (2003) The molecular basis of water transport in the brain. Nat Rev Neurosci 4:991–1001PubMed
Anderson CM, Swanson RA (2000) Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32:1–14PubMed
Anderson TR, Jarvis CR, Biedermann AJ, Molnar C, Andrew RD (2005) Blocking the anoxic depolarization protects without functional compromise following simulated stroke in cortical brain slices. J Neurophysiol 93:963–979PubMed
Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA (2007) Physiological evidence that pyramidal neurons lack functional water channels. Cereb Cortex 17:787–802PubMed
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215PubMed
Atkinson SJ, Hosford MA, Molitoris BA (2004) Mechanism of actin polymerization in cellular ATP depletion. J Biol Chem 279:5194–5199PubMed
Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA (2010) Glial and neuronal control of brain blood flow. Nature 468:232–243PubMedPubMedCentral
Back T, Ginsberg MD, Dietrich WD, Watson BD (1996) Induction of spreading depression in the ischemic hemisphere following experimental middle cerebral artery occlusion: effect on infarct morphology. J Cereb Blood Flow Metab 16:202–213PubMed
Bao L, Locovei S, Dahl G (2004) Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett 572:65–68PubMed
Bargiotas P, Krenz A, Hormuzdi SG, Ridder DA, Herb A, Barakat W, Penuela S, von Engelhardt J, Monyer H, Schwaninger M (2011) Pannexins in ischemia-induced neurodegeneration. Proc Natl Acad Sci U S A 108:20772–20777PubMedPubMedCentral
Barreto GE, Gonzalez J, Torres Y, Morales L (2011) Astrocytic-neuronal crosstalk: implications for neuroprotection from brain injury. Neurosci Res 71:107–113PubMed
Basarsky TA, Duffy SN, Andrew RD, MacVicar BA (1998) Imaging spreading depression and associated intracellular calcium waves in brain slices. J Neurosci 18:7189–7199PubMed
Ben Ari Y (1989) Effect of glibenclamide, a selective blocker of an ATP-K+ channel, on the anoxic response of hippocampal neurones. Pflugers Arch 414(Suppl 1):S111–S114PubMed
Bindokas VP, Miller RJ (1995) Excitotoxic degeneration is initiated at non-random sites in cultured rat cerebellar neurons. J Neurosci 15:6999–7011PubMed
Related posts:
 General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
 Ionic Signaling in Physiology and Pathophysiology of Astroglia
Ionic Signaling in Physiology and Pathophysiology of Astroglia
 Enteric Glial Cells: Implications in Gut Pathology
Enteric Glial Cells: Implications in Gut Pathology
 Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
 Novel Therapeutic Approaches to Malignant Gliomas
Novel Therapeutic Approaches to Malignant Gliomas
 Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


