Antibodies to intracellular and cell-membrane antigens in patients with limbic encephalitis. (A) Sagittal section of rat hippocampus immunolabeled with anti-Hu antibodies. (B, C) Consecutive sections of hippocampus immunolabeled with Kv 1.2 antibodies to voltage-gated potassium channels (B) and a novel neuropil antibody (C). (D) The area in the rectangle in (C) at higher magnification. (E) Reactivity of another neuropil antibody in a consecutive section of the same region. Note the difference between (A) and (B–D); the anti-Hu antibody (A) reacts with intracellular antigens (Hu), while the other antibodies react with areas of the neuropil that are rich in dendrites and synapses but spare the neuronal cell bodies. In all panels, the asterisks are placed in the same region (neurons of the dentate gyrus) to allow comparison between reactivities. Sections counterstained with hematoxylin.
In terms of pathophysiology, more is known about certain syndromes than others. In most cases, antibodies appear to cause down-regulation of their target antigen by antigen internalization; complement-mediated destruction may also play a role in specific syndromes [11]. Neuropathology shows variable T- and B-cell as well as plasma cell infiltrates, though less severe than in pathology of intracellular antibody-associated syndromes, with less cell death [11].
In general, cell surface antibody-mediated syndromes respond well to immunotherapy and tend to be associated with better outcomes than intracellular antibody-associated syndromes [12]. They are also less likely to be associated with an underlying neoplasm [13]. Depending on the specific syndrome, the frequency of associated cancers can vary from as much as 56% in some series of anti-NMDAR to just 3% of anti-GlyR encephalitis [10]. The most common associated tumors include ovarian teratomas, thymomas, breast and lung (particularly SCLC) cancers in those patients with an associated neoplasm [10, 14]. Some syndromes are often associated with specific tumors (e.g., ovarian teratoma in NMDA), which will be discussed in detail below.
Demographics and clinical presentation
Although rare, cell surface antibody-mediated syndromes are thought to be at least five times more frequent than traditional intracellular antibody-related syndromes [5]. They also represent a significant percentage of cases of encephalitis: in a recent study in England, anti-NMDAR or VGKC (LGI1 or CASPR-2) encephalitis represented 8% of all encephalitis cases and 38% of cases identified as having an immune-mediated cause (the remainder were largely classified under the ADEM heading) [15]; an ICU-based study found that approximately 20% of a cohort of 31 patients with encephalitis over a 4-year period had NMDAR antibodies [16]. The incidence of these conditions is likely greatly under-reported. For example, of all patients 30 years or younger referred to the California Encephalitis Project with encephalitis of unclear etiology over a 3.5 year period, anti-NMDAR encephalitis was the single most common identified cause [17]. Representing 40.5% of cases where an etiology was identified, anti-NMDAR encephalitis was more than four times as frequent as HSV-1, VZV, or WNV [17]. It is likely that other antibody-mediated syndromes were among the remaining undiagnosed patients in this and other encephalitis cohorts.
Clinically, cell-surface antibody-mediated syndromes tend to present more acutely than intracellular Ab syndromes, with an average course of less than 12 weeks prior to presentation, and patients quickly reach their clinical nadir [14]. Patients may describe a preceding infectious illness (particularly well-described in NMDAR). Antibody-mediated encephalitis tends to affect women more than men; anti-NMDAR encephalitis is particularly common in younger individuals, including children [10, 14].
Patients may present with limbic encephalitis, other psychiatric and behavioral disturbances, and other neurological signs and symptoms, including neuromyotonia, dysautonomia, ataxia, and dyskinesias. The clinical syndrome of limbic encephalitis, characterized by subacute onset of episodic memory impairment, behavioral and personality changes, disorientation, and agitation, and, frequently, seizures and sleep disturbance, implicates dysfunction in the mesial temporal lobes and amygdala and often orbitofrontal cortex. It was previously thought of as a paraneoplastic syndrome [1] but is now recognized, particularly in the cell-surface antibody-mediated syndromes, as frequently non-paraneoplastic.
Diagnostics and treatment
The diagnostic work-up for patients with suspected cell-surface antibody-mediated syndromes is similar to that for patients with intracellular antibodies (Figure 22.2). The clinician must exclude other inflammatory, infectious, toxic/metabolic, malignant, and vascular etiologies that could be accounting for the symptoms, evaluating specifically for demyelination, SLE, Sjögren’s, sarcoidosis, neuro-Behçet, primary CNS angiitis, and celiac disease; viral, spirochete, prion protein, fungal or mycobacterial disease; thyrotoxicosis, B1 or B12 deficiency, vitamin E deficiency, or heavy metal toxicity; primary malignancy or diffuse metastatic disease; and a shower of embolic infarcts or PRES. We recommend a broad serum and CSF work-up to exclude the above, as well as sending CSF for IgG index, oligoclonal bands, and cytology with flow cytometry. EEG and MRI can be helpful both to exclude other processes and to identify characteristic findings associated with antibody-mediated disease. Both serum and CSF should be sent for broad antibody panels including both common intracellular and cell-surface antibodies. Because patients may have multiple antibodies, syndrome presentations may not be classic for a given antibody, and our understanding of the phenotype of individual antibody syndromes is still evolving, we recommend sending a panel rather than a selected group of individual tests. Particularly if a potentially paraneoplastic antibody is identified, one should pursue a thorough screen for occult malignancy. The clinician may also consider brain biopsy in cases with rapid progression or in which a thorough work-up is unrevealing.
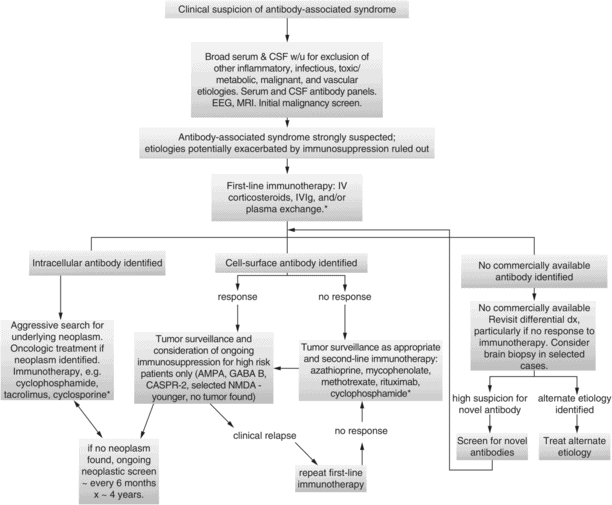
Treatment flow chart. This diagram provides a recommended general approach to the diagnostic work-up and treatment of antibody-associated encephalitis. Our practice, if there is a high suspicion for antibody-associated syndrome, and etiologies (such as infection, lymphoma, etc.) potentially exacerbated by immunosuppression have been ruled out, is to treat patients empirically with first-line immunotherapy while awaiting results of paraneoplastic screen, which can take weeks to return.
In cell surface antibody-mediated syndromes, CSF often shows a moderate lymphocytic pleocytosis, elevated IgG index, and unique oligoclonal bands, though less consistently than in intracellular antibody-associated syndromes [14]. EEG most commonly shows non-specific slowing but is associated with more specific findings or with epileptiform discharges and seizures in specific syndromes, as below [18].
Imaging findings are highly variable, but the most common findings include T2 hyperintensity or atrophy in the medial temporal lobes including amygdala and hippocampus, particularly in patients with limbic encephalitis. There may also be findings of extralimbic cortical involvement as well as basal ganglia, thalamus, brainstem, and cerebellum (Figure 22.3). Imaging findings cannot distinguish between paraneoplastic vs. non-paraneoplastic, or intracellular antibody-associated vs. cell-surface antibody-mediated syndromes; additionally, imaging may be entirely normal in some cases, particularly in early stages of disease. MRI is perhaps most helpful to exclude stroke and other vascular lesions, malignancy, demyelinating lesions, and atypical infections, and it can also help differentiate autoimmune encephalitis from CJD [19].
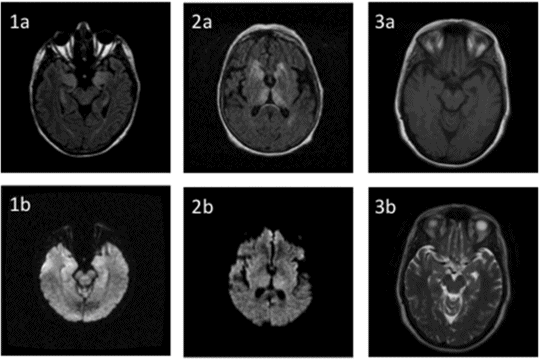
Imaging findings in autoimmune antibody-associated encephalitis. 1. 66-year-old female patient with anti-VGKC (presumed LGI-1) encephalitis. The FLAIR image (1a) shows hyperintensity of bilateral hippocampi as well as left amygdala, with research protocol diffusion tensor imaging sequences suggesting reduced diffusion in the cortex of the bilateral temporal lobes and possible insular involvement (1b). ADC sequence was negative in this patient. 2. 68-year-old female patient with anti-CV2 encephalitis. FLAIR imaging (2a) shows marked hyperintensity in caudate heads and posteromedial thalami, as well as right temporal periventricular white matter, but diffusion imaging was unremarkable (2b). 3. 47-year-old female patient with anti-AMPA receptor encephalitis. T1-weighted imaging shows subtle left hippocampal atrophy (3a). There are no abnormalities on T2-weighted imaging (3b).
If an antibody associated with a cell-surface protein returns positive, or if a cell-surface antibody-mediated syndrome remains a strong diagnostic possibility, and conditions that might be exacerbated by immunosuppression have been excluded, we generally recommend empirically starting immunotherapy. Antibody tests may take a long time to return, many cell surface antibodies are not yet part of standard panels, and these syndromes often respond very well to treatment. The goal of treatment in cell-surface antibody-mediated syndromes is to eliminate pathogenic antibodies and suppress the pathogenic inflammatory process. Typical first-line acute treatments include high-dose IV corticosteroids (e.g., 1 g methylprednisolone daily for 5 days), IVIg (with a standard course of 2 grams per kg delivered over five sessions), and plasma exchange. In most cases, patients improve significantly within weeks after starting treatment, with some exceptions (anti-NMDAR encephalitis may respond more slowly). When a tumor is present, it should be treated aggressively; most patients show an excellent response to tumor removal and oncologic therapy. Sometimes acute therapies are all that is needed, but when the clinical response is incomplete or for syndromes with high risk of relapse, maintenance immunosuppressive therapies should be considered. These currently include oral glucocorticoids, mycophenolate mofetil, azathioprine, methotrexate, rituximab, and pulse cyclophosphamide [20]. Please see the excellent review by McKeon for dosing and monitoring recommendations [21].
In many cases, even patients requiring second-line therapy are able to discontinue immunotherapy in 1 to 2 years without relapse, implying a monophasic course of illness, although there are though notable exceptions, such as anti-AMPAR encephalitis, which is associated with frequent relapses. Although it is clear that patients who do not receive immunotherapy during their initial presentation are more likely to relapse, at least in anti-NMDAR encephalitis [22], when and whether immunotherapy should be used solely to prevent relapse (e.g., in an anti-AMPAR encephalitis patient who has recovered from her initial episode) is under debate. Some clinicians choose to trend serum and (if feasible) CSF antibody titers to assess for treatment response and in hopes of identifying an impending relapse earlier. At a minimum it is imperative to follow these patients closely and consider treatment at the earliest sign of relapse. Some patients, particularly with VGKC-complex-associated disorders, can relapse quickly, over a matter of days. At our center, we follow both the patient’s clinical course as well as his or her antibody titers, although there are insufficient data to support treating based on rising titers alone.
Description of individual cell surface antibody-mediated syndromes
Below, we describe each of the major cell surface antibody-mediated syndromes, discussing epidemiology, pathophysiology, classic clinical presentation, and spectrum of disease, paraneoplastic association, response to therapy, and clinical outcomes. (See Table 22.1 for a summary of the key characteristics of each syndrome, including the number of reported cases.) Disorders are presented in order of most to least commonly diagnosed.
| Cell surface antibody-mediated syndromes with behavioral symptoms | Clinical features | Lab, imaging, and electrophysiology | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Antibody | Number of cases* | Tumor association | Percent with tumor | Age (median) | Sex | Behavioral symptoms | Other neurological and medical symptoms | Clinical course | Serum | CSF | MRI | EEG |
| Anti-NMDAR | > 500 | Ovarian teratoma | 9–56% | 23 mo–76y (19) | F>>M | Psychiatric symptoms, language dysfunction including mutism, memory disorders | Seizures, dyskinesias, autonomic instability and coma | Most do well with early immunotherapy and tumor removal; non-paraneoplastic cases more difficult to treat and 20–25% may relapse | Other antibodies present in ~10% (ANA, TPO) | Inflammatory in 94%; pleocytosis 91%; IT synthesis in > 60% | Abnormal in 50% with transient cortical/subcortical T2 hyper intensities | Abnormal in 88%, with rhythmic monomorphic delta/theta slowing and electrographic seizures in some patients |
| VGKC complex: anti-LGI-1 | >600 VGKCc; at least 188 confirmed LGI-1 | Lung, thymus (rare) | < 20% | 30–80 (60) | M > = F | Limbic encephalitis with amnesia, executive dysfunction, seizures, psychiatric symptoms | Faciobrachial dystonic seizures early in clinical course (40%); hyponatremia (60%) | Monophasic, may not require ongoing immunosuppression | Other antibodies present in about 10% (ANA, TPO, GAD65) | Inflammatory in 41% (pleocytosis 41%); infrequent IT synthesis | Abnormal in about 84% with mesial temporal T2 hyperintensities | May show diffuse mild slowing, bilateral frontal slowing, temporal sharps, or be normal; faciobrachial dystonic seizures may be accompanied by 2–4 Hz spike-wave activity |
| VGKC complex: anti-CASPR2 | > 600 VGKCc; at least 90 confirmed CASPR-2 | Thymoma, rarely SCLC or other | < 20% | 46–77 (60) | M >>F | Limbic encephalitis | Morvan’s syndrome /neuromyotonia, peripheral nerve hyperexcitability | May be treatment-responsive or improve spontaneously, patients with tumor do worse | Other antibodies present in about 20% (MuSK, AChR, GAD65) | Inflammatory in 25%, limited information regarding IT synthesis | Abnormal in about 40% (usually those with encephalitis) with medial temporal T2 hyperintensities | May be normal or show mild diffuse or focal slowing &/or temporal sharps/ epileptiform discharges |
| Anti-GABA B | 62 | SCLC, thymoma | 47–60% | 20–80 (62) | M = F | Limbic encephalitis | Early and prominent seizures, orolingual dyskinesias | Treatment-responsive | Other antibodies present in ~50% (N-type VGCC, GAD65, TPO, SOX1) | Inflammatory in 90%, 80% CSF pleocytosis; frequent IT synthesis | Abnormal in about 2/3 with medial temporal T2 hyperintensities | May be normal, often with generalized slowing or epileptiform discharges |
| Anti-AMPAR | 15 (Dalmau 2009, Bataller 2010, Graus 2010, Wei 2013, and one case at our center) | Lung, breast, thymus | 50–70% | 30–90 (60) | F >>M | Limbic encephalitis, psychiatric symptoms | Seizures, nystagmus | Treatment-responsive, frequent relapses (50–60%) | Other antibodies present in ~60% (ANA, GAD65, cardiolipin, VGCC, SOX1, CRMP5, TPO) | Inflammatory in 90%, pleocytosis 90%; frequent IT synthesis | Abnormal in about 90% with mesial temporal T2 hyperintensities | Often abnormal with frontal or diffuse slow waves, theta activity, frontal or temporal sharps |
| Anti-GABA A | 6 (Petit-Pedrol 2014) | None to date | n/a | 3–63 (22) | 5M, 1F | Memory, cognitive deficits and behavioral changes. Depression, psychosis including hallucinations, mutism | Refractory status or EPC in all patients in initial series. Chorea, ataxia, opsoclonus, dystonic movements, progressive hemiparesis | Partial recovery in most but significant morbidity/mortality associated with status epilepticus | Other antibodies present in 3/6 patients (TPO, GAD65, GABAB R) | Inflammatory in 5/6 of original series with pleocytosis in 4 and IT synthesis in 2 | Abnormal in 6/6 of original series with multifocal non-enhancing T2 abnormalities | EEG with seizures in all, PLEDs in 2 |
| Anti-DPPX | 4 (Boronat 2013) | None to date | n/a | 45–76 | 2M, 2F | Limbic encephalitis, psychiatric symptoms | Myoclonus, tremor, seizures, prodromal diarrhea in 75% | Responsive to immunotherapy with frequent relapses | Other antibodies present in 2/3 tested (ANA, ssDNA) | All with CSF pleocytosis and IT synthesis in initial series | Patchy T2 hyperintensities in 1/4 of initial series; no clear associated patterns yet identified | May be normal or may show diffuse slowing or PLEDs |
| Anti-mGluR5 | 3 (Carr 1982, Lancaster 2011) | Hodgkin lymphoma | 100% | 15, 15, 46 | 1M, 2F | memory loss, cognitive deficits, depression/anxiety, hallucinations | myoclonic jerks (one patient), seizures | Excellent recovery with treatment of Hodgkin’s in all 3 cases; one patient also received corticosteroids | Unknown | Inflammatory in 2/2 with lymphocytic pleocytosis; IT synthesis in 1/2 | Abnormal in 2/2 with patchy T2 hyperintensities including mesial temporal in one and B parietooccipital diffusion restriction in the other | Unknown, with clinical seizures in 2 patients |
* references given for syndromes with < 50 published cases.
Anti-NMDA receptor encephalitis
Background and pathophysiology
Since its initial description by Dalmau et al. in 2007 [23], N-methyl-D-aspartate receptor antibody (anti-NMDAR) encephalitis has vaulted to neurologic and public attention, with over 500 cases reported to date in the medical literature [14] and a popular book, Brain on Fire, about one patient’s experience, capturing the public imagination [24]. Anti-NMDAR encephalitis is likely among the most common forms of antibody-associated encephalitis: in a retrospective study, NMDAR antibodies were identified in 1% of 18–35-year-old patients admitted to one ICU [25]. Anti-NMDAR encephalitis was recently named the most common cause of encephalitis in patients under 30 years old in the California Encephalitis Project, identified more than four times as frequently as HSV, WNV, and VZV over a 3.5-year period [17].
NMDAR is a glutamatergic ligand-gated and voltage-dependent ion channel with a critical role in synaptic plasticity and learning via its role in long-term potentiation. In anti-NMDAR encephalitis, antibodies are directed against the NR1a subunit of the receptor. Based on in vivo and in vitro studies, these antibodies have proven to be pathogenic. Dalmau et al. showed that surface NMDAR clustering in rat hippocampal neurons decreased dramatically after being cultured with patient antibodies, and returned to baseline after removal of antibodies [26]. Hughes et al. demonstrated that this rapid and reversible effect was caused by antibody-mediated capping and internalization, that this effect was independent of complement activation, and that the degree of internalization correlated with CSF and serum antibody titers [27]. The antibodies appear to be secreted by plasma cells which infiltrate the CNS [28], disrupt a critical interaction between NMDAR and Ephrin-B2 receptors, then displace NMDARs from the synapse into the extrasynaptic space, where they are then cross-linked and internalized [29]. Based on animal studies, the effect of this internalization appears to be pathologically enhanced activity of glutamatergic pathways [30], potentially via inhibition of NMDAR on GABAergic interneurons, which express particularly high concentrations of NMDAR [31], although direct effects of patients’ antibodies on GABAergic neurons have not been studied. The presence of NMDAR antibodies has been shown in animal studies to suppress induction of LTP [29, 32].
Demographics and clinical presentation
The syndrome most often affects children or young women and is often associated with ovarian teratoma in adult women, particularly those of African descent; teratoma is found in up to 50% of all cases over age 18 [20, 33]. All examined teratoma cases in the Dalmau series expressed NMDAR. Rarely, anti-NMDA encephalitis is associated with tumors other than teratoma (approximately 2% of patients in a large Dalmau series) [20]. Four of eight patients in a pediatric series had other autoimmune markers such as positive ANA or thyroperoxidase, suggesting a predisposition to autoimmune disease [34]; cases following vaccination have also been described [20]. Recent work also suggests that some patients with NMDAR antibody encephalitis may previously or concurrently develop clinical and neuroimaging abnormalities such as optic neuritis and longitudinally extensive myelitis with positive NMO or MOG antibodies suggestive of neuromyelitis optica (NMO) spectrum disorder [35].
Anti-NMDAR encephalitis has a striking clinical presentation: the first symptoms are often psychiatric, and many patients are initially admitted to psychiatric wards for delusions, hyper-religiosity, paranoia, mania, or anxiety. Patients also frequently suffer from sleep dysfunction, memory deficits, and decreased verbal output, echolalia, or even mutism. Approximately 70% report a prodrome of flu-like symptoms, GI complaints, or headaches within 2 weeks of psychiatric symptom onset [20]. Within days after their first cognitive symptoms, patients may develop characteristic movement disorders, including oral–facial dyskinesias, but also choreoathetosis, dystonia, rigidity, and opisthotonus. Early in the course of illness, patients often develop seizures; these may be a presenting symptom (i.e., prior to psychiatric or other symptoms), particularly in men [36]. Seizures can be confused with movement disorders and vice versa, complicating management.
Over a period of days to weeks, many patients develop more pronounced abnormal movements accompanied by dysautonomia and often decreased level of consciousness. Central hypoventilation is common, with many patients becoming comatose and/or requiring intubation, resulting in prolonged stays in intensive care. Autonomic storms can become severe; rarely, prolonged cardiac pauses require pacemaker placement. Near their clinical nadir, patients have been noted to exhibit dissociative states similar to those caused by NMDAR antagonist anesthetics [20]. The severity of clinical illness does not appear to correlate with the presence or absence of teratoma. As patients clinically improve, they tend to recover in stages, first from coma and dysautonomia, then from seizures and dyskinetic movements. Psychiatric and behavioral symptoms tend to linger longest and may persist to some degree for months to years despite treatment.
Children tend to present similarly to adults, although psychiatric symptoms tend towards irritability, tantrums, and violent behavior rather than psychosis and may be less likely to come to medical attention [20]. Later in the course of illness, dysautonomia and hypoventilation are less common in children [34]. The dyskinetic subset of patients with encephalitis lethargica, a pediatric encephalitis syndrome which characteristically presents with disordered sleep, neuropsychiatric symptoms, and movement disorders, likely represents a pediatric form of NMDAR antibody encephalitis [37].
Diagnostics
CSF is typically inflammatory, with a lymphocytic pleocytosis and, in 60% of patients, intrathecal synthesis of oligoclonal bands. CSF abnormalities are present on initial presentation in approximately 80% of patients and develop later in the course of illness in most of the remainder [26]. In the experience of one group, high titers are more common in patients with teratoma and in those with poor outcome; the same group finds that NMDA receptor antibody testing is significantly more sensitive in CSF than serum (100% vs. 85.6%) [38], although another laboratory finds serum at 1:20 dilution to be slightly more sensitive than undiluted CSF [33]. Serum titers appear to be higher than CSF titers for most patients [33, 39]. Given the higher sensitivity of CSF, and that serum titers have been shown to be positive in up to 10% of the population of blood donors [40], we strongly recommend obtaining both CSF and serum samples for analysis.
Imaging is frequently unrevealing but approximately 50% of patients will have variable cortical and subcortical T2 hyperintensities on MRI, sometimes with mild enhancement after contrast administration, which may be transient or chronic [20]. Significant atrophy has been reported in patients with refractory disease, including refractory seizures [23, 41]; frontotemporal atrophy and hypoperfusion were also found at 7–12 months after diagnosis in two young women; both women showed significant recovery of cognitive function over time, and both findings had reversed on follow-up imaging at 5–7 years [31].
EEG is nearly always abnormal and characteristically shows rhythmic, monomorphic delta or theta slowing or disorganized activity (seen in 88% of patients in a series by Florance et al. [34]), which does not correlate with clinically observed movement disorders. Some patients also show electrographic seizures, however, with or without clinical correlates, and video EEG monitoring with aggressive treatment of seizure activity is recommended [34]. Rarely, anti-NMDA receptor encephalitis has been associated with complex partial or non-convulsive status epilepticus, which can be very treatment-resistant [42, 43].
Brain biopsies have been performed in several cases and typically show non-specific inflammation, with perivascular lymphocytic cuffing and microglial activation [26].
When anti-NMDAR encephalitis is suspected or diagnosed, a search for ovarian tumor should be pursued promptly. Screening at our center typically consists of pelvic and/or transvaginal ultrasound (in adult patients) followed by MRI of the pelvis if unrevealing. Blind oophorectomy is sometimes pursued when the MRI is normal and patients are extremely ill and at times reveals teratoma, although this can be a very difficult question to navigate medically and ethically in a prospective fashion given the natural history of prolonged critical illness in a subset of patients with NMDAR encephalitis even in the absence of teratoma [43]. Patients without identified neoplasms at the time of acute illness should also receive periodic screening for ovarian teratoma for at least 2 years (with some guidelines recommending screening for as long as 4 years [41]) as anti-NMDAR encephalitis can predate detectable tumors [37].
Treatment
Although there are no established protocols for treatment of anti-NMDAR encephalitis, first-line therapy generally consists of high-dose IV corticosteroids, IVIg, and/or plasma exchange. It is our practice to generally begin with steroids (IV methylprednisolone 1 g/day for 5 days or equivalent orally) typically with the addition of IVIg (generally 2 grams/kg divided over 5 days). Some groups recommend weekly therapy consisting of either 1 g IV methylprednisolone or 0.4 g/kg IVIg once weekly for 6–8 weeks following the initial course [18]. The Dalmau group recommends concurrent methylprednisolone and IVIg (using the same dosing and duration as above) [20]. In one large cohort, response to immunotherapy tended to be slower than in other cell-surface Ab syndromes, with a 53% response to first-line immunotherapy within 4 weeks in a recent large cohort; of those who did not respond to first-line treatment, second-line immunotherapy improved outcomes in an additional 57% [44]. Patients who undergo removal of an identified tumor respond more quickly and dramatically to first-line immunotherapy than other patients, with substantial improvement in 80% of those with tumor removal, as opposed to 48% of those without, after first-line immunotherapy [20]. Second-line immunotherapy, if required, typically consists of rituximab and/or cyclophosphamide, and may be continued for up to 1 year after completion of first-line immunotherapy in order to minimize the risk of relapse [20, 34, 44, 45]. In severe cases, there may be a benefit to using both agents concurrently [20]. Please see the excellent review by McKeon for detailed recommendations on second-line immunotherapy dosing and monitoring [21].
After treatment, 75% of patients recover completely or with only mild deficits [26]. Based on data for 360 patients with at least 6-month follow-up, Dalmau et al. report an estimated mortality of 4%, with almost all deaths occurring in the ICU [26]. Relapses of illness can occur over months to years in 12–25% of patients, most commonly those without known neoplasms [26, 33, 34, 44], but most relapses are less severe than initial episodes [44]. Although both serum and CSF antibody titers diminish highly significantly with treatment, one large study shows that relapses are more strongly associated with an increase in CSF than in serum titers [38]. It may be helpful to periodically screen the serum and CSF to assess the effects of treatment and predict potential relapses [20]. Though no clear guideline on timing of follow-up screening has been established, we recommend screening every 6–12 months for 2–4 years. As noted above, we additionally recommend periodic screening for teratoma for 2–4 years following the initial episode if none is identified at the time of acute illness.
Expanded clinical spectrum
Recent work has also shown the presence of NMDAR antibodies in both CSF and serum in children and adults with relapsing symptoms after an initial episode of HSV-1 encephalitis, which is non-responsive to acyclovir [46, 47]. NMDAR antibodies are especially likely to be found in relapsing children with chorea [48, 49]. Such symptoms may be responsive to immunotherapy, and based on initial studies immunotherapy appears to be safe in these cases [48, 49].
The question of false positives
The interpretation of NMDAR antibodies is complicated by a large recent study that identified NMDAR antibodies in the serum of 10.0% of a sample of 4,236 individuals, including random anonymous blood donors (“healthy controls”) and individuals with psychiatric and neurological comorbidities, with levels approaching 20% of stroke and ALS cases. This study unfortunately did not examine CSF, so it is not clear if any of these would have had positive CSF titers [40]. Generally, when considering NMDAR encephalitis, serum and CSF should be assessed for antibodies. This study also raises the important point that antibody testing is most useful in patients with a high pretest probability of having an antibody-mediated syndrome, and that positive antibodies found in a healthy subject are not, in themselves, indicative of disease.
The relationship between anti-NMDAR encephalitis, psychosis, and ketamine
It is intriguing to compare the clinical syndrome of anti-NMDAR encephalitis to current models of NMDAR dysfunction in schizophrenia and in anti-NMDAR anesthetics such as ketamine and phencycline. The work of John Krystal and colleagues has highlighted the similarities between patients with early schizophrenia and the effects of ketamine on healthy individuals [50]. Dalmau et al. note that the clinical phenotype of anti-NMDAR encephalitis overlaps heavily with these syndromes, and further that the comatose and dissociative states found in patients receiving high doses of ketamine bear a striking resemblance to the most severe stage of anti-NMDAR encephalitis [20]. Interestingly, new work provides evidence for increased prevalence of anti-NMDAR in patients with admissions for acute schizophrenia, though antibodies were not IgG directed against the NR1a subunit, as in anti-NMDAR encephalitis, but IgA, IgM, or antibodies directed against both NR1a and NR2b [51]. IgA antibodies directed against the NR1a subunit were also identified in seven of a series of 24 patients with a rapidly progressive dementia syndrome, affecting not only NMDAR density but also altering other key synaptic proteins [52]. These patients generally had atrophy on neuroimaging but normal CSF and EEG findings. Whether IgA antibodies against the NMDAR represent a separate primary autoimmune syndrome, exist in association with a broader variety of pathogenic antibodies, or represent a secondary immune response to a primary neurodegenerative process is not yet clear. The finding of positive serum NMDAR antibodies in non-classic disorders must be interpreted with caution in light of recent findings in one study that about 9.4% patients with schizophrenia, 19.2% of stroke patients, and 8.5% of healthy anonymized blood donors test positive for serum NMDAR antibodies [40]. On the other hand, new research also demonstrates that many de novo mutations found in schizophrenic patients affect glutamatergic function, including mutations affecting proteins associated with the NMDA receptor [53].
Studies in the psychiatric literature have demonstrated decreased NMDAR availability in unmedicated schizophrenics in vivo using SPECT [54] as well as a block in what is likely, based on animal studies [55], an NMDA-mediated response to theta stimulation of visual cortex as measured by VEP magnitude in schizophrenic patients [56]. Single-cell recordings in non-human primates as well as neuroimaging studies in both healthy individuals on ketamine and schizophrenics show attenuated DLPFC activity during working memory tasks, potentially related to NMDAR antagonism [57–59]. These indirect techniques of assessing NMDAR functioning in vivo have not yet been applied to anti-NMDAR encephalitis, and might represent non-invasive ways to measure disease progression and predict relapse in the future.
X was a healthy 18-year-old woman and solid B student who had done well and enjoyed the first half of her senior year of high school. In the spring semester, however, her family began to notice some unusual behaviors. Although the family had always been occasional churchgoers, they were never particularly religious, and so they were surprised when X began carrying a Bible everywhere she went and admonishing her siblings by quoting scripture. Several days later, X began loudly singing hymns at the dinner table. She would often refuse to eat at all, or had specific requests, only accepting macaroni and cheese for days at a time. One night, about 10 days after symptom onset, she woke her parents, screaming that the devil was in her room; her family brought her to the emergency room.
Her initial work-up included an unremarkable serum screen for rheumatologic, toxic, and infectious processes. CSF showed four WBCs (all lymphocytes), 38 RBCs, glucose of 69 (serum 100) mg/dl, and protein of 56 mg/dl; oligoclonal bands and IgG index were not performed. Viral studies, cultures, and VDRL were negative. Brain MRI was concerning for FLAIR hyperintensities in the bilateral mesial temporal lobes and insula. A CT abdomen/pelvis was read as concerning for a left-sided cystic adnexal structure; MRI of the pelvis was read as a likely simple ovarian cyst.
During her hospitalization, X continued to have marked hyper-religiosity, holding her Bible at all times, responding to questions with chanting and loud singing, and expressing frequent concerns that devils and angels were in her room. She had episodes of acute fearfulness, one of which was accompanied by urinary incontinence, during which she maintained awareness. Initially, she ate some meals with gentle coaxing, but after several days she stopped eating completely. She began to develop tongue-thrusting movements as well as periods of agitation accompanied by bicycling movements; both of these types of episode were felt concerning for seizure. She was intermittently tachycardic. Three days into her admission, she was transferred to the ICU after becoming acutely more poorly responsive. Her mental status fluctuated to the point that she was sometimes unarousable to sternal rub. Her neurological exam, apart from her mental status, remained non-focal.
After a repeat LP with additional infectious and neoplastic diagnostics, X received a three-day course of high-dose IV steroids, followed by 2 g IVIg given over five daily doses. Unfortunately, her condition did not improve. She remained in the ICU with fluctuating but declining mental status. She was intubated for inability to protect her airway. She continued to have episodic tachycardia. Continuous EEG monitoring showed rhythmic delta activity, which did not correlate with her tongue-thrusting and bicycling movements. Serum and CSF NMDAR antibodies were positive. Five sessions of plasmapheresis yielded no improvement. Although second-line immunotherapy was discussed, the decision was made to proceed next with oophorectomy of the left ovary despite MRI read of likely simple cyst. Pathology of the left ovary confirmed a teratoma. In the days following oophorectomy, X was weaned off the ventilator and her autonomic instability improved dramatically. She began speaking and interacting and her episodes of tongue-thrusting and bicycling became less frequent. About 5 weeks after symptom onset, she celebrated her nineteenth birthday in the ICU and was able to laugh, interact with family members, and enjoy a piece of birthday cake. She was transitioned to the ward and discharged less than one week later. In follow-up, her unusual movements have resolved, as have her hyperreligiosity and delusions. She has returned to school but finds it more difficult than before to pay attention in class and concentrate on her homework. She struggles to maintain a passing average. She has required no further immunotherapy.
Disorders with antibodies to the voltage-gated potassium channel complex (VGKCc): anti-LGI-1 and –CASPR-2 encephalitis
The voltage-gated potassium channel complex
Voltage-gated potassium channel complex (VGKCc) antibody encephalitis, which commonly presents as limbic encephalitis, neuromyotonia (or Isaacs’ syndrome), or Morvan’s syndrome, was the first immunotherapy-responsive surface antibody syndrome to be well characterized [60, 61]. It continues to be a relatively commonly reported syndrome, with more than 600 cases reported as of 2012, mostly in the UK [14]. Although initial studies suggested that the syndrome was related to antibodies to the voltage-gated potassium channel (VGKC) [62], more recent studies have shown that antibodies target proteins that are tightly complexed with VGKC (Figures 22.3 and 22.4) [63, 64]. In 2010, in a group of VGKCc limbic encephalitis patients, the leucine-rich glioma-inactivated 1 (LGI-1) protein was identified as the key autoantigen by immunoprecipitation. The following year, another group identified contactin-associated protein 2 (CASPR-2) autoantigen as the pathogenic antibody in a group of VGKCc patients with limbic encephalitis and peripheral nerve hyperexcitability, and this has since been confirmed to be the autoantigen in other cases of neuromyotonia that had been previously attributed to anti-VGKC [64–66]. Rarely, antibodies can also target contactin 2, another part of the VGKCc. Many of these patients are also positive for LGI-1, CASPR-2, or other VGKC-related antibodies; whether contactin-2 plays a role in a primary autoantibody syndrome is not yet clear [64]. There are likely pathogenic VGKCc antibody targets which have not yet been identified, as some patients with a clear syndrome of limbic encephalitis test positive for VGKCc antibodies, but not for antibodies to LGI-1, CASPR-2, or contactin [67].
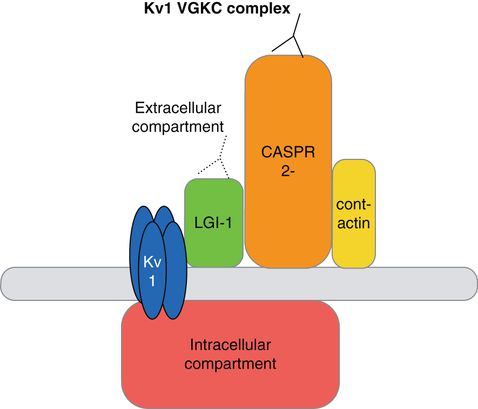
Kv1 voltage-gated potassium channel complex schematic. Antibodies previously thought to bind to the VGKC appear to bind instead to elements of the VGKC complex, including LGI-1 and CASPR-2. Both LGI-1 and CASPR-2 may be present at the presynaptic VGKCc; antibodies may also bind to LGI-1 and CASPR-2 when they are not complexed with the VGKC.
Patients with VGKCc antibodies may present with limbic encephalitis, seizures, and peripheral nerve hyperexcitability, including Morvan’s syndrome, or a combination of these. VGKCc antibodies may also be detected incidentally in patients with a low suspicion for CNS autoimmunity, though LGI-1 and CASPR2 are not usually positive in such cases [67]. According to one study, low-positive levels of VGKCc antibodies (100–400 pM) appear only to be pathogenic in cases of peripheral nerve hyperexcitability and, in those cases, are associated with tumors in 12.5% of 32; high-positive levels (> 400 pM) were more likely to be clinically relevant and more likely to accompany a clinical presentation of limbic encephalitis [67]. The typical presentations for LGI-1 and CASPR-2 antibodies vary, as described below, with LGI-1 patients more likely to present with limbic encephalitis and seizures and CASPR-2 patients more likely to present with Morvan’s syndrome. Although the classic behavioral symptoms are those associated with limbic encephalitis, VGKCc antibody patients, particularly those with LGI-1, may present with rapidly progressive dementia, which can mimic FTD [68] or strongly resemble Jakob–Creutzfeldt disease [3, 69]. On the other hand, several cases have been reported in which patients with proven prion disease also test positive for VGKCc antibodies. In one case, a patient with Jakob–Creutzfeldt disease diagnosed definitively on pathology was found to be positive for VGKCc antibodies (603.5 pM); testing for IgG binding to LGI-1, CASPR-2, and contactin unfortunately was not performed so it is not clear if this VGKCc antibody was a false positive [70]. In another case, a patient with pathology-proven Gerstmann–Sträussler–Scheinker (GSS) disease due to a novel prion protein gene mutation was also found to be strongly positive for VGKCc antibodies (4159 pM), though notably the patient’s IgG did not bind to LGI-1, CASPR-2, or contactin (but did bind to the surface of hippocampal neurons in culture) [71]. These cases underscore not only the potential clinical similarity of patients presenting with prion disease and VGKCc antibodies, but also the importance of distinguishing VGKCc antibodies incidental to a general autoimmune response from pathogenic VGKCc antibodies. As the tests for these associated proteins are more specific than the VGKCc RIA because they involve expression of only that protein in cell lines, determining if antibodies to specific VGKCc proteins, such as LGI-1 or CASPR-2, are present may be helpful in determining whether the finding is an artifact of general autoimmune response or more specific, as the tests for these associated proteins involve expression of only that protein in cell lines, and are therefore more specific than the VGKCc RIA.
Anti-LGI-1 encephalitis
Background and pathophysiology
LGI-1 is a secreted neuronal protein that interacts with both the presynaptic protein ADAM23 and the postsynaptic protein ADAM22, and is thought to play a role in the control of synaptic AMPA receptors. It also has been associated with autosomal dominant lateral temporal lobe epilepsy. In rat models, using serum from patients with limbic encephalitis, it appears to reversibly reduce synaptic AMPA receptor clustering [72], potentially leading to decreased AMPA receptor functioning in GABAergic neurons and thereby causing pathologic excitability [65].
Demographics and clinical presentation
Patients with anti-LGI-1 encephalitis, unlike anti-NMDAR or -AMPAR patients, are more commonly male (~65%), and usually present with limbic encephalitis [5]. In a large subset of cases (~40%)[65], encephalitis is often preceded by episodes of brief tonic–myoclonic movements, which may represent tonic seizures [73]. These movements might involve a limb, the face or head, or trunk, can be unilateral or bilateral and might be somewhat diffuse. Sometimes there is brief memory loss or confusion with the episodes. Some investigators have named these movements faciobrachial dystonic seizures (FBDS), although in many cases there is no evidence of seizure on EEG [74, 75]. Thus far these episodes appear to be relatively specific for VGKCc disease and especially LGI-1. Prompt recognition of these and treatment with immunotherapy seems to decrease the likelihood of progression to encephalitis and more widespread seizures [74, 75]. These movements can be mistaken for myoclonus, leading to further diagnostic confusion with prion disease [3]; 82% of LGI-1 patients have clinical seizures, often temporal in focality [65]. Up to 60% of patients will also present with hyponatremia [64, 65], often attributed to SIADH and potentially related to LGI-1 expression in the hypothalamus or kidney [65]. We recently identified a bradycardia syndrome as the first symptom in three of our 17 LGI-1 patients [76]. Underlying neoplasms are uncommon in LGI-1 encephalitis (< 20%), but when present typically involve lung or thymus.
Although VGKCc disorders are commonly thought of as involving prominent memory deficits, other marked cognitive deficits are frequently present. In recent work from our group, 12 patients with LGI-1 who had comprehensive neuropsychological testing overall demonstrated significant deficits in memory and executive function. Some also had deficits in tests of linguistic functioning, but visuospatial functioning generally was spared [77].
Diagnostics and treatment
CSF is inflammatory in just 41% of patients; oligoclonal bands are seen rarely [60, 64]. The majority (56–84%) of patients have T2 hyperintensities in the medial temporal lobes (MTLs) on MRI [5, 64]. In chronic phases, MRI may also show atrophy, particularly in the mesial temporal lobes, even after clinical recovery [11]. Neuropathology, when obtained, in some cases shows lymphocyte infiltration as well as prominent immunoglobulin and complement fixation [11] whereas in others shows features of encephalitis including microgliomatosis and neuronophagia and in the white matter severe astrocytic gliosis [3]. Patients respond well to immunotherapies with significant improvement usually noted in a matter of weeks; some patients even improve spontaneously [62, 64]. However, some patients suffer significant neurological disability and the question of how aggressive to be with immunosuppressive therapy in this syndrome is an area of active study. Relapse is relatively rare, occurring in only 18% of patients in one landmark series [65].
Anti-CASPR-2 encephalitis
Background and pathophysiology
A member of the neurexin superfamily, CASPR-2 plays a key role in the distribution of VGKCs at juxtaparanodes along myelinated axons [78]. Mutations have been associated with schizophrenia, psychosis, seizures, and intellectual delay [79–81].
Demographics and clinical presentation
Encephalitis associated with antibodies to CASPR-2 is much less common than LGI-1 encephalitis and represents less than 10% of VGKC-complex encephalitis. As with LGI-1 encephalitis, there appears to be male predominance (~85%), and patients typically present with a combination of cognitive symptoms (often limbic encephalitis), seizures, and peripheral nervous symptoms (hyperexcitability, allodynia, neuromyotonia) [64, 66]. Patients may also present with Morvan’s syndrome, characterized by insomnia, psychosis, dysautonomia (e.g., hyperhidrosis), and peripheral nerve hyperexcitability which can manifest as neuromyotonia or pain [82]. Peripheral nerve involvement may be secondary to antibody-mediated attack on CASPR-2 in peripheral nerves [66]. Some patients present with progressive bulbar weakness and are initially found to have myasthenia gravis, with positive AChR or MuSK antibodies [66].
Diagnostics and treatment
Of patients with CASPR-2 and limbic encephalitis, about 40% have T2 hyperintensities in the medial temporal lobe. CSF is only abnormal in about 25% of patients [5, 66]. Although in patients with Morvan’s syndrome approximately 40% have associated neoplasms, often thymoma (and at times a history of associated myasthenia gravis), among CASPR-2 cases with limbic encephalitis, associated tumors are less common (probably < 20%) [64, 66]. Patients found to have tumors have a poorer prognosis than those without [64].
Patients with CASPR-2 antibodies may not respond to immunotherapy as well as patients with AMPA, GABA, or LGI-1 antibodies [64, 66] and, as for anti-NMDAR (without tumor) and anti-AMPAR encephalitis, treatment should be aggressive [5], such as considering chronic immunosuppression with azathioprine or mycophenolate mofetil [21].
Anti-tag-1/contactin-2 encephalitis
Tag-1/contactin-2 is a cell adhesion molecule that binds with and supports CASPR-2 [78]. In a series of patients who were positive for VGKCc antibodies, five of 96 were positive for contactin-2; four of these were also positive for other VGKCc antibodies [64]. Whether anti-tag-1/contactin-2 defines its own clinical syndrome remains to be determined.
Anti-GABABR encephalitis
Background and pathophysiology
A recently described syndrome, anti-GABA B receptor (anti-GABA-BR) encephalitis is likely the most common cause of limbic encephalitis in patients with SCLC previously considered “seronegative” because of the absence of anti-Hu or other antibodies [83, 84]. A metabotropic G-protein-coupled receptor, GABA B plays a crucial role in the control of neuronal excitability by presynaptic inhibition [85], and is distributed throughout the CNS but particularly densely in the hippocampus, cerebellum, and thalamus [86]. In animal studies, absence of the GABA B receptor is associated with spontaneous seizures and memory impairment [87], and receptor polymorphisms are associated with temporal epilepsy in humans [88]. Although to date there is no definitive evidence of in vitro antibody pathogenicity in anti-GABA B encephalitis, patients’ antibodies bind GABA B in live rat neurons [89].
Demographics and clinical presentation
In the original series of 15 patients, described by Lancaster and colleagues, all presented with limbic encephalitis accompanied by prominent seizures [89]. A more recent series of 20 patients by Hoftberger and colleagues looked for anti-GABA antibodies in a large pool of individuals with presumed autoimmune or paraneoplastic encephalitis symptoms of unclear etiology. Although 17 of the 20 GABABR positive patients presented with limbic encephalitis and seizures (which were difficult to control in five), three patients presented with unusual symptoms including: 1. prominent seizures with myoclonic jerks (and no limbic encephalitis); 2. gait ataxia, spasticity, and dysarthria; and 3. opsoclonus–myoclonus syndrome, respectively [7].
Diagnostics and treatment
Unlike many cell-surface antibody-mediated syndromes, anti-GABABR is frequently associated with neoplasm, specifically SCLC, which is found in 50–60% of patients, usually after neurologic symptom onset; paraneoplastic anti-GABABR tends to present at a much older age than patients with non-paraneoplastic disease (mean 67.5 vs. 39 years in the Hoftberger et al. cohort) [7, 89]. As in anti-AMPA encephalitis (below), patients are often found positive for other autoimmune antibodies, perhaps reflecting a diffuse autoimmune response to neoplasm or a general tendency to autoimmunity. Approximately 30–50% of patients have such additional autoantibodies, including SOX1, N-type VGCC, GAD65, TPO, Ri, BRSK2, amphiphysin, and NMDAR [7, 83, 89]. Most patients with onconeuronal antibodies (e.g., SOX1, BRSK2, Ri, or amphiphysin) are found to have underlying SCLC [7].
During inpatient evaluation of anti-GABABR patients, EEG often shows epileptiform discharges or general slowing. CSF is usually inflammatory, and unique oligoclonal bands are frequent. In 45–67% of patients, MRI shows T2 hyperintensities in the medial temporal lobe [7, 89].
Although most patients’ neurological symptoms respond quickly and at least partially to first-line immunotherapy, mortality in reported cases is relatively high despite neurological response, usually related to progression of underlying SCLC (and, in one case, refractory status epilepticus) [7, 83]. Because of the frequency of underlying lung cancer, patients diagnosed with anti-GABABR encephalitis, similar to anti-AMPAR patients, should have regular neoplastic screening for about 4 years following diagnosis, as discussed elsewhere for paraneoplastic syndromes [90].
M. was a healthy 74-year-old woman when she started to notice involuntary twitching movements of the right eyelid. Six months later, the twitching became stronger and began to involve more of her right face as well as her arm, often causing her to drop items. The movements might happen up to a dozen times a day and, a month later, began to affect her left arm as well. Around the time the twitching became more noticeable, her family began to notice personality changes: she was more easygoing than usual and lost her interest in politics. By the time her left arm became involved, family found she was less talkative, showed less interest in family members’ lives, and no longer worked in her garden or on the crossword puzzle. Her long-standing insomnia became significantly worse and her handwriting became smaller. A month later, she began having gait ataxia with frequent falls and had difficulty grooming and dressing, and she moved in with her daughter for assistance. She was diagnosed with Jakob–Creutzfeldt disease (JCD) and referred to our clinic for an urgent appointment and possible enrollment in a treatment trial. She appeared encephalopathic with a waxing and waning mental status. Her gait was ataxic and she had occasional simultaneous jerking or spasm of the right arm and right face, which lasted a few seconds, longer than a typical myoclonic jerk. We suspected autoimmune encephalitis, particularly VGKCc, and admitted her to our hospital for further work-up.
On admission, she was drowsy, flat, and provided only brief answers to questions. MMSE was 18/30. Her exam was also notable for paratonia, diffuse mild atrophy, and occasional myoclonus in the left arm, with low-amplitude taps in all extremities. Her reflexes were brisk and symmetric with pathologic spread but down-going toes; gait was wide-based, halting, and unsteady. An extensive serum and CSF work-up was notable for hyponatremia to 120 mmol/L throughout her hospitalization, with urine labs consistent with SIADH, as well as CSF with 0 WBCs, protein of 44 mg/dL, normal IgG index of 0.5, and four unique oligoclonal bands. EEG showed diffuse slowing. MRI brain showed increased T2/FLAIR and DWI signal (without clear ADC map correlate to suggest restricted diffusion) in the left frontal lobe and anterior cingulate and underlying white matter and bilateral hippocampi; the MRI was felt to be potentially concerning for JCD. Chest CT showed a small calcified nodule in the left lower lobe and hilar prominence, raising the question of prior granulomatous disease. CT abdomen/pelvis showed a renal mass consistent with her prior imaging and felt to be a possible renal cell carcinoma; PET showed non-specific increased uptake in bilateral hilar region of the lungs, but not in the kidney. Given the concern for possible CJD, brain biopsy was obtained and showed no vacuoles or prion immunostaining, but was, however, notable for astrocytic gliosis in gray matter only without lymphocytic infiltrate. Following biopsy, paraneoplastic screen per the Mayo Clinic returned very elevated for anti-VGKCc, as well as antibodies to the N-type calcium channel.
She was started on a 5-day course of high-dose (1000 mg/day) IV steroids followed by a 3-month oral prednisone taper. MMSE improved to 30/30, but she was still felt to require 24-hour supervision, and returned home with her daughter. In clinic follow-up 1 month later, she continued to be somewhat confused, with family members reporting waxing and waning levels of awareness. She still spoke mostly in short phrases but naming and recall were markedly improved. She had occasional left arm jerking on exam and tended to lean forward when walking, with a stably wide-based gait. She was prescribed a second course of 5 days of high-dose IV steroids. Following steroids, she and her daughter felt that she had returned to her cognitive baseline. She continued to have possible myokymia under the bilateral eyes and did not recall events from the few months surrounding her hospitalization. She remained mildly hyponatremic at 131 mmol/L.
She was readmitted several months later after a generalized tonic clonic seizure, with exam showing ataxic gait and cognitive decline and labs showing worsened hyponatremia. She received a third course of high-dose IV steroids and 2 g IVIg over 5 days, and then began mycophenolate as second-line immunosuppression. She continued to have seizures and was maintained on valproic acid and levetiracetam. Due to concern of the renal mass being a carcinoma and possibly associated with her syndrome, it was resected, turning out to be a benign oncocytoma. Over the following 2–3 years she transitioned to rituximab monotherapy, with cognitive function improving dramatically (most recent MMSE 28/30) to the point at which she was beating her children in chess again. She continued to have profound apathy, but refused treatment for this, and some short-term memory problems. Aricept was started with apparent dramatic improvement in her short-term memory. Both VGKCc and N-type VGCC antibodies have recently become negative. Over about 4 years, she has received four rounds of rituximab (either 500 or 1000 mg/dose), but none in the past year and a half, with clinical stability.
Anti-AMPA receptor encephalitis
Background and pathophysiology
A rare cause of limbic encephalitis or acute psychiatric symptoms, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antibody encephalitis was first described in a case series of ten patients in 2009 [91], with 15 cases described to date to our knowledge [92–94]. The initial case series demonstrated that the patients had antibodies against the GluR1 or GluR2 subunits of the AMPA receptor. Applying patients’ antibodies to cultures of live rat hippocampal neurons caused internalization of AMPA receptors, decreasing GluR2+ AMPAR synaptic clustering as well as overall AMPA cluster density [91], and supporting the idea that the antibodies are pathogenic.
The AMPA receptor (AMPAR), a glutamatergic ligand-gated ion channel, is the greatest mediator of fast excitatory activity in the brain. Via the processes of long-term potentiation and depression, it is heavily involved mechanistically in learning and memory [95] and also plays a key role in seizure generation and spread [96]. AMPA receptors with the highest density of GluR1 and GluR2 subunits are found in the hippocampal CA1-2, subiculum, cerebellum, caudate and putamen, as well as cortex [91].
Demographics and clinical presentation
Based on the limited data available, patients with AMPAR antibody encephalitis typically present in the fourth to eighth decade of life, with a median age of approximately 60 years; 90% are women. Most develop a subacute limbic encephalitis syndrome with prominent memory disturbance (see clinical vignette). Patients may alternately present with primarily psychiatric symptoms, including apathy, agitation, aggressive behavior, and hallucinations [92, 93]. Some patients may have subtle findings of incoordination or motor impairment, potentially related to involvement of AMPA receptors in the basal ganglia or cerebellum. A substantial minority of patients (40% in the Lai et al. series) also developed seizures [91]. Notably, approximately half of the 15 described patients also have a history or antibody profile suggestive of other systemic autoimmune disorders, such as thyroid disease, DM, Raynaud’s syndrome, a positive ANA, or dsDNA, or other forms of antibody-associated autoimmune encephalitis, including anti-GAD, anti-CV2, and anti-VGCC.
Diagnostics and treatment
A majority of cases (ten of the 15 cases known, of whom seven had a cancer diagnosis prior to presenting episode of LE) have been paraneoplastic, including cancers of the lung, breast, and thymus, and in these cases tumors have been shown to express GluR1 and GluR2 subunits matching the antibodies found in those patients’ CSF [91]. CSF is typically inflammatory with a lymphocytic pleocytosis; MRI shows T2 hyperintensities in the medial temporal lobes in most cases thus far [91].
In one case report, a pregnant patient with LE in the setting of AMPA receptor antibodies had a fulminant course, requiring intubation during the acute period, with a CSF lymphocytic eosinophilic pleocytosis, T2 hyperintensities in bilateral medial temporal lobe, insula, and caudate, and the acute development of cortical atrophy [94]; such fulminant neurological decline and cortical atrophy, however, have not otherwise been reported in the setting of this syndrome. We have seen relatively rapid cortical atrophy, over about 1 year or so, however (Geschwind et al., unpublished).
In general, patients respond well to immunotherapy (in combination with oncotherapy when a neoplasm is present), though residual symptoms, including fatigue and memory, and attentional difficulties, may persist. Unfortunately, per the available literature, AMPAR encephalitis also appears to be associated with a high risk of relapse (14 relapses in 5/9 patients over up to 101 months in the original series) [91]. In our experience in our autoimmune encephalopathy clinic and research program, in the setting of relapse, most patients respond only partially to treatment. For this reason, just as in other cell-surface antibody-mediated encephalopathies without identified tumors, if patients continue to be symptomatic, some clinicians recommend chronic or long-term immunosuppression with mycophenolate mofetil, azathioprine or possibly agents such as rituximab, particularly if the antibody levels remain elevated. Because of the high incidence of associated cancer, we recommend adhering to the European Federation guidelines for neoplastic screening in typical paraneoplastic syndromes, which suggest screening about every 6 months for a period of 4 years [90]. This is a conservative approach, however, given limited information from the few cases reported.
S. was a 42-year-old, well-regarded executive with a large multinational corporation when she began having some unexpected difficulties at work. She was having trouble finding words while writing or speaking. Tasks that would have been easy for her to manage previously took much more time, and she often found herself staring blankly at her computer screen. A few weeks after she began to notice these changes, a coworker called her husband to report that she had repeated the same story several times during lunch. Five days later, she visited her local hospital for a routine screening test, but after the appointment could not find her car despite searching for half an hour. At that point her husband was concerned and brought her urgently to see her primary doctor. She was admitted to her local hospital, where basic labs were unremarkable and LP was bland with 0 WBCs and protein 28 mg/dl (no IgG index or oligoclonal bands were sent at this time); viral PCRs and VDRL returned negative. MRI showed subtle T2 hyperintensities in the bilateral temporal lobes and EEG was concerning for epileptiform activity in the same areas, though without clinical episodes concerning for seizures.
During her hospitalization, S.’s memory continued to decline; her conversations with her husband consisted entirely of the same questions and answers repeated over and over. She was discharged with plans for further outpatient work-up, but stayed up all night at home packing for a trip, unpacking, and repeating the process. She began to seem more confused and tired; she stopped eating and was disinterested in her children. Her husband brought her back to the hospital, where she became subdued and then increasingly somnolent. She was transferred to our medical center where repeat CSF analysis revealed a lymphocytic pleocytosis (WBC 19 and 7 on separate occasions), a protein of 28 mg/dl, which rose to 48 (normal 15–45) on a repeat lumbar puncture, as well as two unique oligoclonal bands and an elevated IgG index of 1.2 (normal < 0.7). Viral studies, cultures, and VDRL were again negative. CSF was also sent to the California Encephalitis Project and to the research laboratory of Dr. Joseph Dalmau. A broad serum work-up for rheumatologic, toxic (including heavy metals), and infectious processes that might be contributing to her decline was entirely unremarkable. A neoplastic work-up including CT abdomen/pelvis and full-body PET showed no evidence of malignancy. MRI again showed subtle T2 hyperintensity and enhancement in the bilateral temporal lobes. EEG now showed diffuse slowing but no clinical or electrographic seizures. S. was started on an empiric 5-day trial of high-dose steroids with some improvement in her mental status – her husband recalls that near the end of these 5 days she opened her eyes spontaneously for the first time. Soon after this, research testing in the laboratory of Dr. Josep Dalmau reported AMPA receptor antibodies in the CSF (serum not sent). She began a five-session course of plasmapheresis, after which she began to improve dramatically; she began to track and identify family members and then to speak. She first realized she suffered from significant retrograde amnesia when her sons, whom she recalled as 4-year-old twins, came to visit her and were 13 years old.
She returned home nearly 3 months after her symptoms began and describes a slow process of cognitive recovery, describing mental exhaustion, difficulty focusing, depressed mood, and easy fatigue. She was able to complete activities of daily living (ADLs) but often forgot steps in more complex tasks, such as remembering to turn on the oven when baking. Nearly a year after symptom onset, a serial PET scan showed increased uptake in her breast; she was diagnosed with ductal carcinoma based on biopsy. She was treated with surgical resection, chemotherapy, and radiation and is now in remission. She continues to work on her problems with memory, attention, and fatigue. To this day she has only rare, vague memories of the 9 years prior to her illness.
Related posts:
 The non-fluent/agrammatic variant of primary progressive aphasia
The cognitive neurology of corticobasal degeneration and progressive supranuclear palsy
The non-fluent/agrammatic variant of primary progressive aphasia
The cognitive neurology of corticobasal degeneration and progressive supranuclear palsy
 Amnestic mild cognitive impairment
Amnestic mild cognitive impairment
 The Lewy body dementias
The Lewy body dementias
 Semantic dementia (semantic variant primary progressive aphasia)
Semantic dementia (semantic variant primary progressive aphasia)
 Sleep issues in dementia
Sleep issues in dementia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


