Table 13.2 Autonomic Epilepsies Presenting at Specific Ages
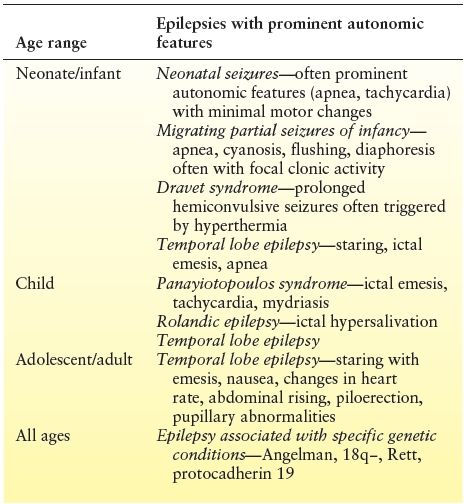
Table 13.3 Nonepileptic Conditions with Prominent Autonomic Features by Age Group at Presentation

The pathophysiology of autonomic changes with seizures most likely involves direct excitation or inhibition of the neocortical and limbic cortices involved at seizure onset and their propagation to structures that constitute the central autonomic network (1,9). This network involves an extensive neural circuitry that extends from the forebrain to the brain stem and includes the insular cortex, amygdala, hypothalamus, periaqueductal gray matter, parabrachial complex, nucleus of the tractus solitaries, and ventrolateral medulla. Through this circuitry, autonomic, visceromotor, neuroendocrine, pain, and behavioral responses are controlled. Inputs to the central autonomic network are multiple including viscerosensory inputs relayed through the nucleus of the tractus solitaries and humoral inputs relayed through the circumventricular organs. Higher-order autonomic control is mediated by the prefrontal and insular cortices and amygdala. Electrical stimulation of these regions results in various autonomic signs. Stimulation of the right anterior insular cortex is reported to elicit tachycardia and pressor responses, whereas bradycardia and depressor responses are reported to occur with left anterior insular stimulation. Stimulation of the medial prefrontal cortex results in changes in blood pressure, heart rate, and gastrointestinal motility. The amygdala and bed nucleus of the stria terminalis are responsible for integrated autonomic and motor responses to emotion.
Children appear to be particularly vulnerable to autonomic seizures and, in particular, ictal emesis. In adults, ictal vomiting is rare and is usually seen later in a seizure once consciousness is impaired and after onset of other temporal lobe symptoms. Conversely, ictal vomiting in children is common and usually seen at seizure onset with intact consciousness and without preceding focal cortical symptoms. It has been suggested that there is a maturation-related susceptibility for the central autonomic network, with children being more vulnerable to emetic disturbances (10). Furthermore, in children, central autonomic networks may have a lower threshold to epileptogenic activation than those producing focal cortical semiology. Regardless of the localization of onset of ictal discharge, the lower threshold autonomic centers may be activated initially, with consequent autonomic manifestations. Other higher threshold cortical regions that produce focal semiology such as motor, sensory, or other symptoms are only activated later and if the ictal discharge reaches a certain threshold.
Specific Epilepsies and Seizures with Prominent Autonomic Features
Neonatal Seizures
Seizures are among the most common neurologic disorders in neonates, with an incidence of 1 to 3 per 1000 live births. Most seizures in neonates are reflective of underlying brain pathology, although the list of potential etiologies is diverse. Seizures in the newborn are usually focal and often have subtle semiology with eye deviation, staring, blinking, nystagmus, mouthing, or chewing. Autonomic features are also very common, including apnea, abnormal respiratory patterns, hiccups, tachycardia, and/or hypertension (11). Profound ictal apnea may be the only manifestation of neonatal seizures arising from either the temporal or the occipital regions (12,13), although associated heart rate changes are rare in ictal apnea (14). The cause of the frequent association between neonatal seizures and autonomic features has not been well studied; however, connections between the posterior limbic cortex, the temporal lobe, and the midbrain respiratory centers have been postulated (13).
Epilepsy of Infancy with Migrating Focal Seizures
This rare but devastating syndrome presents in three distinct phases (15). In the first few weeks to months of life, focal motor seizures, which evolve to bilateral convulsive events, occur. Autonomic features are very common and include apnea, flushing, and cyanosis. The second phase is seen between 1 and 12 months and is characterized by a marked increase in seizure frequency, often to a point where seizures are nearly continuous. Prominent ictal autonomic features persist. The EEG at this time shows the typical migrating and multifocal pattern of ictal epileptiform discharges. During the third phase, which typically is seen between 1 and 5 years of age, the frequency of seizures markedly reduces, although clusters can still occur during intercurrent illnesses. The etiology of many cases is unknown, although a variety of genetic causes have been reported, including SCN1A and KCNT1 mutations. Therapy is challenging, with some response reported with bromides, a combination of stiripentol and clonazepam, levetiracetam, or rufinamide (16). Two patients with profound ictal apnea and bilateral temporal involvement experienced significant improvement with addition of acetazolamide (17), possibly due to either cessation of bitemporal seizures or induction of metabolic acidosis that resulted in stimulation of central chemoreceptors.
Dravet Syndrome
Dravet syndrome is a devastating epilepsy syndrome that begins in the first 18 months of life with recurrent prolonged seizures, which are intractable to medical therapy. Initial seizures are often hemiconvulsive or evolving to bilateral convulsive activity. By age 2 years, other seizure types including myoclonic jerks, absences, focal seizures, and drop attacks emerge. Focal seizures with prominent autonomic features occur in over half of patients, with pallor, cyanosis, respiratory changes, and drooling, often associated with eye deviation, myoclonus, or focal motor activity. A significant provoking factor for seizures is hyperthermia due to intercurrent illness, postimmunization fever, physical exercise, warm baths, or elevated ambient temperatures. While caregivers frequently report disturbed body temperature regulation, red or bluish hands/feet, alteration in sweating, pupillary dilation, facial or chest flushing, tachycardia at rest, or slow gastric emptying, few studies have carefully evaluated autonomic function in these children.
Approximately 80% of children have a demonstrable mutation in the SCN1A gene. The risk of SUDEP in this cohort is high, with an approximate annual incidence of 0.6% (18). SCN1A is primarily a neuronal gene; however, several studies have shown that the product of this gene, Nav1.1, is present in the heart in several animal species. It appears important in normal activity of the sinoatrial node, control of heart rate, and heart rate variability (19). A small number of studies have suggested a relative predominance of adrenergic tone in Dravet syndrome, as evidenced by reduced heart rate variability, and P-, QT-, and QTc wave dispersion (20, 21). Such changes may predispose to an increased risk of tachyarrhythmias and possibly explain the higher risk of SUDEP, although further studies are needed.
Panayiotopoulos Syndrome
Autonomic seizures and autonomic status epilepticus are core features of Panayiotopoulos syndrome, a common electroclinical syndrome that affects preschool or early school-aged children (7). Seizures frequently arise from sleep with a collection of autonomic symptoms and mainly emesis, with or without vomiting, tachycardia, syncope-like manifestations, unilateral eye deviation, and progressive altered awareness, which may progress to hemiconvulsions.
EEG studies in children with Panayiotopoulos syndrome typically reveal multiple foci, often with an occipital predominance. Using magnetoencephalography, Saito et al. noted that younger patients had spikes in the calcarine, parietooccipital, or rolandic regions, as opposed to older children in whom frontal foci were seen (22). These results are in keeping with a maturation-related cortical hyperactivity in this syndrome, which may result in activation of the lower-threshold central autonomic network.
Benign Epilepsy with Centrotemporal Spikes
Benign epilepsy with centrotemporal spikes is the most common focal epilepsy seen in children and typically presents with diurnal focal seizures affecting the ipsilateral lower face and occasional ipsilateral upper extremity and/or nocturnal generalized seizures. One of the characteristic clinical features is profound ictal hypersalivation, which is usually seen with ipsilateral lower facial motor activity and dysarthria. Rarely, this syndrome may present with prolonged hypersalivation and oromotor and speech disturbances suggestive of an anterior opercular syndrome.
Temporal Lobe Epilepsy
Temporal lobe epilepsy often presents with autonomic features, particularly in children. “Abdominal epilepsy” may be seen, characterized by periumbilical, colicky abdominal pain, often accompanied by headache, dizziness, syncope, temporary loss of vision, and impaired consciousness. The duration of abdominal pain is typically no longer than 15 minutes and may be associated with sweating, borborygmi, salivation, and flatus.
Rarely, profound apnea attacks may be the sole manifestation of temporal lobe epilepsy, and this is most commonly seen in preschool children and infants. However, a case of SUDEP in a 30-year-old woman with temporal lobe epilepsy, who had previously documented ictal apnea, suggests that this autonomic disturbance may be a significant risk factor for SUDEP (23). Ictal apnea appears most likely with spread of temporal lobe epilepsy to the contralateral hemisphere (24).
The most common autonomic change seen in temporal lobe epilepsy is ictal tachycardia, which is present in up to 98% of childhood temporal lobe seizures. Ictal bradycardia is less frequent, and while it may be due to activation of the left temporal and insular cortex, a careful study found that it does not have consistent lateralization but rather appears after seizure activity becomes bilateral (25). Cardiac pacing should be considered in cases of ictal asystole.
Autonomic features suggestive of specific lateralization in temporal lobe seizures include both abdominal auras with vomiting and orgasmic auras, which both lateralize to the nondominant temporal lobe. Other symptoms that are not specific to lateralization and that may also be seen with nontemporal foci include postictal cough, flushing, pallor, sweating, piloerection, mydriasis, and miosis.
Epilepsy Associated with Specific Genetic Conditions
Ictal apnea appears to be particularly common in many epilepsies due to specific genetic conditions including protocadherin 19 mutations, 18q− syndrome, 1p36 deletion syndrome, and trisomy 18. Accurate diagnosis of the underlying etiology of autonomic symptoms in these genetic disorders can be quite challenging, as many have autonomic disturbances unrelated to seizures as well. Girls with Rett syndrome typically have peripheral vasomotor disturbances and breathing dysfunction. Additionally, cardiac testing has shown an exaggerated increase in heart rate response to breath holding and prolonged QTc suggestive of cardiorespiratory dysregulation. In 18q−, breathing abnormalities including hyperventilation or apnea crises are common. Video-EEG monitoring may be needed to determine if such symptoms have epileptiform correlate.
SUDDEN UNEXPECTED DEATH IN EPILEPSY
SUDEP is the most important direct epilepsy-related cause of death. It is defined as a sudden, unexpected, witnessed or unwitnessed, nontraumatic, and nondrowning death in a patient with epilepsy. This may or may not occur in the setting of an epileptic seizure and excludes deaths resulting from status epilepticus. If the above criteria are met and an autopsy is performed, revealing no toxicologic or anatomical cause of death, the death is termed “definite SUDEP.” “Probable SUDEP” is diagnosed if the above criteria are met, but an autopsy is not performed. When no autopsy is performed, insufficient evidence relating to the cause of death exists, and SUDEP cannot be excluded, a diagnosis of “possible SUDEP” can be made.
SUDEP has an estimated incidence of 1.8 per 1000 patient-years (26). However, higher incidences (3 to 9/1000 patient-years) have been reported with intractable epilepsy (27). While SUDEP is rarer in the pediatric population, it is still estimated to be between 1 and 2 per 10,000 patient-years (28).
Based on previous studies, the most consistently identified risk factor for SUDEP is poorly controlled generalized tonic–clonic seizures (29–33). Other, less-robust risk factors include antiepileptic drug polytherapy, developmental delay/intellectual disability, nocturnal seizures, young age at seizure onset, and longer duration of epilepsy (29–33). The use of specific drugs such as carbamazepine or lamotrigine has been suggested as potential risk factor however, recent careful studies have refuted these findings (34,35).
Prevention of SUDEP should involve increased efforts to decrease the frequency of generalized tonic–clonic seizures. Furthermore, supervision at night may be associated with a decreased risk of SUDEP (29).
SUDEP AND PERI-ICTAL AUTONOMIC DYSFUNCTION
While the exact pathophysiology of SUDEP has yet to be fully elucidated, ictally mediated autonomic dysfunction is most likely responsible. Such dysfunction may involve cardiac disturbances, including peri-ictal tachycardia, bradycardia, asystole, repolarization (QTc) anomalies, and reduced heart rate variability. Potentially fatal decreases in cerebral oxygenation could also result from peri-ictal hypoxemia and respiratory suppression. Recently, the contribution of postictal generalized EEG suppression to SUDEP has been studied. Through retrospective review of electrophysiologic data from people who later died of SUDEP and prospective studies of those at greatest risk, we continue to learn more about the mechanisms responsible for sudden death.
Peri-Ictal Tachycardia
Given its high prevalence, there is great interest in the potential role of peri-ictal tachycardia in SUDEP. Peri-ictal tachycardia (often defined as a heart rate greater than the 98th percentile for age) is the most commonly observed seizure-related autonomic disturbance, occurring during and/or after a majority of recorded seizures (36). Based on stimulation of the human insular cortex, Oppenheimer et al. (37) hypothesized that tachycardia is more likely to result from seizures lateralizing to the right hemisphere. However, peri-ictal tachycardia has more consistently been documented in association with mesial temporal lobe onset and seizure generalization (36).
Some studies of patients who later died of SUDEP suggest a link between peri-ictal tachycardia and resulting death. When comparing maximal ictal heart rates in patients who later died of definite/probable SUDEP to controls with refractory focal-onset seizures, significantly higher values were found in the SUDEP group (38). In addition, significant postmortem fibrotic changes in the deep and subendocardial myocardium have been detected in SUDEP cases versus controls, which could be the result of myocardial ischemia from repetitive seizures (39). Sympathetic overactivity may be responsible for such tachycardia and cause transient dilatation of ventricular walls and left ventricular dysfunction. If such stress-induced cardiomyopathy was severe enough to diminish cardiac output, oxygen supply during periods of significant stress might be compromised enough to result in death.
Peri-Ictal Bradycardia and Asystole
Related posts:
 Epidemiologic Aspects of Epilepsy
Application of Electroencephalography in the Intensive Care Setting
Psychogenic Nonepileptic Seizures
Carbamazepine, Oxcarbazepine, and Eslicarbazepine
Corpus Callosotomy and Multiple Subpial Transection
Continuous Spike-and-Wave During Sleep Including Landau–Kleffner Syndrome
Epidemiologic Aspects of Epilepsy
Application of Electroencephalography in the Intensive Care Setting
Psychogenic Nonepileptic Seizures
Carbamazepine, Oxcarbazepine, and Eslicarbazepine
Corpus Callosotomy and Multiple Subpial Transection
Continuous Spike-and-Wave During Sleep Including Landau–Kleffner Syndrome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


