Basal Ganglia and Brainstem Anatomy and Physiology
Karen Gale
Mark Proctor
Jana Velíšková
Astrid Nehlig
Introduction
Seizure activity does not spread diffusely throughout the brain, but propagates along specific anatomic pathways. The particular pathways responsible for triggering and propagating a seizure determine the nature of the seizure and the pattern of the motor manifestations that accompany it. Although seizures may result from any of a broad array of brain insults, the motor concomitants of the seizures fall into only a few categories of remarkably stereotyped behaviors. This suggests that the anatomic substrates of propagated convulsive seizures in the intact organism may likewise fall into a few major categories or systems. In this chapter, some of the subcortical anatomic substrates of seizure propagation are reviewed. In addition, we identify certain anatomic circuits that are responsible for regulating seizure susceptibility and act as common modulators for multiple seizure-generating circuits. The pathways within the basal ganglia and brainstem will be the main focus of this chapter, with passing reference to other subcortical structures with which these systems interconnect.
Before considering particular anatomic substrates, it is important to recognize that brain regions and pathways may participate in the seizure process in distinctive respects. Some of the roles that a brain area or pathway may play include:
Epileptogenic “trigger” area. A site in the brain that is capable of evoking propagated seizure activity upon focal electrical or chemical stimulation. The trigger area is analogous to a “switch.” It sets in motion the events that result in a seizure, but it is not necessarily one of the first areas to exhibit ictal activity during seizure development.
Epileptogenic “target” area. A site in the brain that is especially vulnerable to the development of ictal activity, either by virtue of its anatomic inputs (e.g., from a “trigger” area) or by virtue of its intrinsic circuitry. This area would be one of the first areas to exhibit a pattern of ictal discharge during the initial development of a seizure, but it is not necessarily a source for seizure propagation.
Pathways involved in propagation of the seizure. These include pathways connecting “trigger” areas and “target” areas, pathways creating positive or negative feedback circuits, pathways allowing the seizure to spread to additional brain loci, and commissural pathways allowing bilateral spread.
Gating inputs. Neural inputs to “trigger” or “target” areas that modulate the excitability of these regions. Changes in the activity of the gating inputs alter seizure threshold in a predictable fashion. These inputs alone are not necessarily capable of inducing seizures.
The first three of these categories are generally specific to seizure type. Gating inputs, on the other hand, exert a common influence on many different seizure types. Brainstem circuitry is most often associated with gating functions, whereas forebrain circuitry has typically been studied for trigger and target components.101,102,104
Overview of Anatomy
Basal Ganglia Circuits
The basal ganglia are a collection of nuclei situated in the forebrain and midbrain that include the caudate nucleus and putamen (together referred to as “striatum”), globus pallidus, entopeduncular nucleus (equivalent to the internal segment of the globus pallidus), subthalamic nucleus, and substantia nigra. The striatum represents the “receiving” end of the system, and the internal segment of the globus pallidus (entopeduncular nucleus in rat) and substantia nigra represent the output end of the system. Three major relays mediate the impact of the basal ganglia on behavior: thalamus, intermediate and deep layers of superior colliculus, and the pedunculopontine area of the caudal mesencephalic tegmentum. Some of the circuitry that interconnects these regions is illustrated in FIGURE 1.
Brainstem Projections
The midbrain and pontine regions of the brainstem contain the cell groups from which the ascending serotonin-containing pathways originate (dorsal and median raphe nuclei) and from which the noradrenergic pathways originate (locus ceruleus and nearby cell groups). Important cholinergic projections also derive from specific cell groups in the caudal midbrain and rostral pons. These monoaminergic and cholinergic projections reach widespread regions of the diencephalon and telencephalon in the forebrain (see Fig. 2). These projections play a critical role in the regulation of sleep–waking states, arousal, and attention, in addition to influencing seizure susceptibility.
Independence of Seizure Generating Mechanisms in Forebrain and Brainstem
Forebrain Mechanisms
It has been demonstrated in the cat that complete transections disconnecting the forebrain from the brainstem do not interfere with the electroencephalographic seizure discharge recorded from the forebrain in response to pentylenetetrazol.173 More recently, in the rat, Browning et al. found that electroencephalographic seizure activity evoked focally from the area tempestas, an epileptogenic site in the deep anterior piriform cortex, was not impaired by complete transections of the brainstem at the pre-, mid- or postcollicular level.24 These observations indicate
that circuitry within the forebrain is sufficient to initiate, propagate, and sustain bilateral seizures in the absence of neural connections with the midbrain or hindbrain.
that circuitry within the forebrain is sufficient to initiate, propagate, and sustain bilateral seizures in the absence of neural connections with the midbrain or hindbrain.
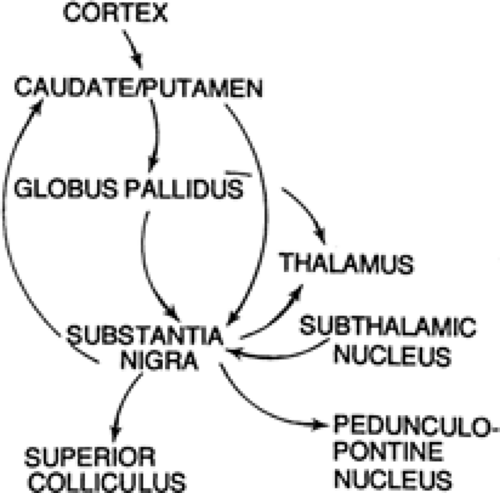 FIGURE 1. Basal ganglia and some of their interconnections, inputs, and outputs. |
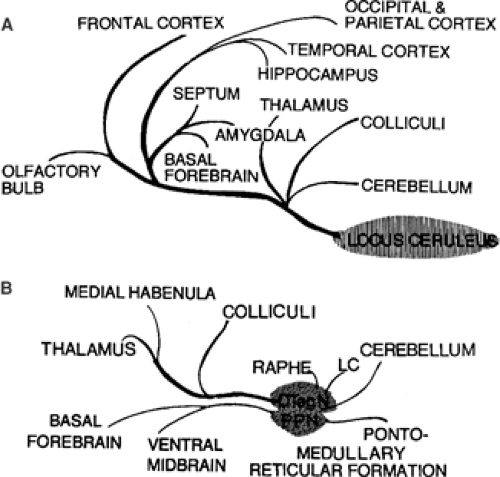 FIGURE 2. A: Noradrenergic projections ascending from locus ceruleus. B: Cholingergic projections ascending from dorsal tegmental nucleus (DTegN). LC, locus ceruleus; PPN, pedunculopontine nucleus. |
Brainstem Seizures That Do Not Depend on Connections with Forebrain
Although forebrain regions are crucial in many forms of epilepsy, the brainstem has multiple roles in both the triggering and propagation of certain types of seizures. A “brainstem” seizure model has been suggested by Gioanni et al. with regard to the quaking mutant mouse. They found that focal stimulation of multiple brainstem regions can induce tonic–clonic seizures in these animals.121 Likewise, brainstem substrates are critical in the induction of audiogenic seizures in susceptible animals, and massive ablation of forebrain structures does not prevent these convulsions.117 Brainstem structures are also sufficient for tonic convulsions evoked by maximal electroshock or chemoconvulsants.28 These observations indicate that, for certain seizure types, the brainstem contains the trigger areas and target areas as well as the pathways required for the full expression of the seizures.
Several animal models of convulsive seizures frequently include explosive running and bouncing characterized by vigorous clonic movements of all four limbs; a tonic component involving the flexion/extension of all limbs, and loss of righting reflex often follows. This convulsive pattern is produced by systemic administration of chemoconvulsant drugs such as pentylenetetrazol, picrotoxin, and bicuculline, as well as by acoustic stimulation of animals that are prone to audiogenic seizures.25,103 These types of seizures have been shown to depend on neural substrates in the brainstem, and do not require the integrity of the forebrain for either their initiation, development, or expression.25,28
Inferior Colliculus as a Site of Seizure Initiation
Stimulation of Inferior Colliculus
The inferior colliculus is a brainstem site from which running/ bouncing clonic seizures can be triggered in rodents. Electrical stimulation of the inferior colliculus in the rat, as well as focal application of drugs that stimulate excitatory amino acid transmission in this site, elicit sudden, explosive bouts of leaping, running, and bouncing on all four limbs.82,94,95,187,194 This behavior resembles that associated with sound-induced (audiogenic) seizures in susceptible rodent strains, and although it can also be evoked by disinhibition of periaqueductal gray neurons,10 it is not dependent on the integrity of the periaqueductal gray for its expression. These inferior colliculus-evoked seizures in rats, which can be induced as early as 3 days of life, have been compared to human neonatal seizures. In fact, McCown and Breese suggest that many neonatal human seizures may actually be initiated in the inferior colliculus.183
The electroencephalographic features of seizures electrically evoked from the inferior colliculus have been well characterized by McCown et al.187 During the wild running behavior, afterdischarges are seen in the region of the inferior colliculus, without abnormalities in the forebrain. Upon repeated stimulation of the inferior colliculus, there is a kindling-like progression to forelimb manifestations (myoclonus or tonic extension); this is associated with both afterdischarges in the region of stimulation and electrical spiking activity in the frontal cortex.
γ-Aminobutyric Acid and Glutamate in the Inferior Colliculus
Blockade of γ-aminobutyric acid (GABA) transmission in the inferior colliculus evokes convulsive effects similar to audiogenic seizures,94,95 increases auditory evoked potentials, and induces susceptibility to audiogenic seizures in otherwise normal rats.11 Faingold et al. found that infusion of baclofen (a GABAB agonist) or gabaculine (a GABA transaminase inhibitor) into the inferior colliculus of genetically epilepsy-prone rats (GEPRs) significantly reduced the severity and incidence of audiogenic seizures;81 muscimol, a GABAA agonist, was almost twice as potent as baclofen at reducing seizures in this model. Similarly, tiagabine, a GABA uptake inhibitor that increases extracellular GABA levels, significantly reduced seizure severity in the GEPR when given either systemically or focally into the inferior colliculus. This occurred in conjunction with a reduction in neuronal firing in the central nucleus of the inferior colliculus.85 Taken together, these observations indicate that GABAergic transmission within the inferior colliculus provides a crucial inhibitory influence on susceptibility to sound-induced seizures.
An essential role for N-methyl-D-aspartate (NMDA) and non-NMDA glutamate transmission in audiogenic seizure initiation was suggested by Faingold et al.84 based on evidence that the NMDA and non-NMDA antagonists were able to block audiogenic seizures in GEPRs when focally applied to the inferior colliculus. Focal application of an inhibitor of glutamate synthesis in the inferior colliculus also blocked audiogenic seizures in the GEPR,86 whereas focal application of glutamate agonists in this region induced running–bouncing seizures in normal rats.82,194
Bicuculline in the inferior colliculus evoked only submaximal seizure activity, whereas NMDA agonists fully reproduced the tonic–clonic seizure seen during audiogenic seizures, suggesting that disinhibition alone, without increased excitation, is not sufficient for full seizure expression in the GEPR.117 In the aftermath of an audiogenic seizure, depletion of excitatory amino acids in the inferior colliculus may contribute to the postictal refractory period.243
Interactions Between Inferior Colliculus and Other Brainstem Structures
Lesions or focal inhibition of inferior colliculus transmission interferes with the generation of sound-evoked seizures not only in the genetically epilepsy-prone rat26,73,83,149,300 but also in DBA/2 mice306 and in rats undergoing alcohol withdrawal.95 Cortical, not central, inferior colliculus nuclei appear to be critical for expression of these seizures.117
The inferior colliculus, as part of the auditory sensory system, is probably a crucial afferent relay station for converting acoustic stimulation into convulsive discharge. Consistent with this view is the reciprocal positive transfer between kindling of audiogenic seizures and electrical kindling of the inferior colliculus140 and the fact that subconvulsant stimulation of this region can render otherwise normal animals susceptible to audiogenic seizures.82,194 Direct chemical or electrical stimulation of this brain region does not typically evoke tonic convulsions in normal rats unless ascending norepinephrine (NE) projections have been compromised by lesions or depletion of NE.31
Detelencephalated rats exhibit enhanced susceptibility to audiogenic seizures in the presence of bicuculline in the inferior colliculus. Therefore, the forebrain structures may normally exert some inhibitory control over these seizures.12 The specific target areas within the brainstem engaged by the inferior colliculus–evoked convulsions have not yet been elucidated, but it is likely that there are critical glutamate-mediated connections in the mesencephalic and pontine tegmentum.193 Mid-collicular knife cuts decrease the wild running component of audiogenic seizures, indicating that inferior colliculus–superior colliculus interconnections are required for some components of audiogenic seizures.245,269 Complete blockade of audiogenic seizures has been obtained with knife cuts that separate the central and external nuclei of the inferior colliculus, suggesting that seizure activity triggered in the central nucleus propagates out of the inferior colliculus via the external nucleus.245,269 Because corticocollicular fibers terminate mainly in the external nucleus, this region may be an important link in a positive feedback loop involving the cortex. In fact, the actual seizure discharge pattern may become organized in the network engaged by the external nucleus, because this nucleus exhibits a low threshold for bursting in disinhibited collicular slices in vitro.233 Pierson and Snyder-Keller233 suggest that a bidirectional excitatory loop between superior colliculus and inferior colliculus may provide for an important oscillatory interaction that sets up the seizure discharge. According to their model, an initial depolarization of external nucleus cells contributes to the epileptiform discharge that develops in the deep superior colliculus and dorsal cortex of the inferior colliculus. The bursting in superior colliculus in turn precedes the synchronous repetitive bursting that occurs in the external nucleus. Thus, the deep superior colliculus is a crucial component of this oscillatory pattern. The participation of the external nucleus and dorsal cortex of inferior colliculus, as well as the deep superior colliculus, was also indicated in vivo by the pattern of c-fos expression associated with audiogenic seizure development.233
A high degree of lateralization is present in the inferior colliculus. Unilateral kindling of the inferior colliculus sufficient to produce running–bouncing seizures and bilateral electrical afterdischarge in the rat, does not alter the de novo kindling rate of the contralateral inferior colliculus. This lateralization is supported by 2-deoxyglucose studies.184 Thus, the progression to running–bouncing seizures is distinct for each side. This contrasts with forebrain seizures in rats; for example, unilateral amygdala kindling facilitates kindling of the contralateral amygdala.185
Brainstem Substrates of Tonic Extensor Convulsions
In rodent models involving maximal or supramaximal convulsive stimuli, tonic extension of the forelimbs or hindlimbs is the typical convulsive endpoint. During the tonic extensor phase of a seizure, the forelimbs are rigid and extended caudally against the ventral surface of the body, while the hindlimbs are rigid and extended horizontally and away from the prone or supine body in a caudal direction. This tonic extensor phase is characteristic of seizures evoked by maximal electroshock or high doses of chemoconvulsants and is usually maintained for 3 to 12 seconds. It is also observed in two strains of genetically epilepsy-prone rats (GEPR 9 rats) and Wistar audiogenic sensitive rats (WAS) in response to acoustic stimulation.220,244,293
As in the case of running–bouncing convulsions, tonic extensor convulsions require the integrity of the brainstem but not the forebrain.28,235 However, the anatomic substrates within the brainstem responsible for generating tonic seizures have not been characterized. Neurons within the reticular formation of the caudal midbrain and pons are likely to be involved, based on the fact that stimulation of this general region triggers tonic extensor convulsions, and lesions within this area can selectively attenuate tonic convulsions.20,29,30,157 Moreover, sustained multiunit activity in the reticular formation correlates with the tonic extensor component of seizures induced by pentylenetetrazol; this activity is not abolished following transections that disconnect the hindbrain from the forebrain at the precollicular level.279
The tonic extensor components of audiogenic and maximal electroshock–induced convulsions are selectively suppressed by lesions of the nucleus reticularis pontis oralis (RPO) without altering clonic seizure activity.25,30 Furthermore, focal injections of a local anesthetic (lidocaine) directly into the RPO reversibly blocked the expression of tonic extensor convulsions.25,30 A similar selectivity for tonic extensor seizure manifestations is seen with lesions of the superior cerebellar peduncles25,30 or removal of the cerebellum.237
Interactions Between Brainstem and Forebrain
Midbrain Regulation of Forebrain Seizure Spread
At the same time that brainstem regions serve to regulate seizure susceptibility in forebrain circuits, they participate in the
development of certain changes that occur in the course of the limbic kindling process. Midsagittal section of the brainstem prior to kindling of the amygdala in rats did not alter the development of kindling at the primary (i.e., first) site but was able to prevent the positive transfer effect at the secondary site (i.e., contralateral amygdala).137 In all intact animals, kindling of the second amygdala proceeded at a rate several-fold faster than the primary site kindling (two to five vs. nine to fourteen stimulations). In contrast, the majority of bisected rats exhibited no enhancement of the secondary-site kindling rate. These observations confirm that interhemispheric connections within the brainstem are not required for amygdala seizure development and, at the same time, suggest their importance for the interhemispheric transfer of susceptibility to kindled seizures. This is supported by studies in feline amygdala kindling, in which bisection of the midline brainstem from the midbrain to pons prevented the positive interhemispheric transfer effect.130,298 It also appears that the midline brainstem participates in symmetrical patterning and electroclinical tonic expression of the kindled seizure. It is possible that the kindling process removes inhibitory influences of specific (as yet unidentified) brainstem regions and that this removal of inhibition contributes to the progressive enhancement of limbic seizure susceptibility. Presumably, brainstem structures homolateral to the primary kindling site must participate in the progression of kindling at that site via intrahemispheric projections. This notion is supported by the observation that unilateral lesions of the midbrain reticular formation markedly reduce susceptibility to seizures evoked by kindling of the homolateral amygdala in cats.299 Apparently, the bisection of the brainstem can prevent the kindling-induced changes from occurring in the contralateral hemisphere, rendering that hemisphere “naive” to the kindling stimulation.
development of certain changes that occur in the course of the limbic kindling process. Midsagittal section of the brainstem prior to kindling of the amygdala in rats did not alter the development of kindling at the primary (i.e., first) site but was able to prevent the positive transfer effect at the secondary site (i.e., contralateral amygdala).137 In all intact animals, kindling of the second amygdala proceeded at a rate several-fold faster than the primary site kindling (two to five vs. nine to fourteen stimulations). In contrast, the majority of bisected rats exhibited no enhancement of the secondary-site kindling rate. These observations confirm that interhemispheric connections within the brainstem are not required for amygdala seizure development and, at the same time, suggest their importance for the interhemispheric transfer of susceptibility to kindled seizures. This is supported by studies in feline amygdala kindling, in which bisection of the midline brainstem from the midbrain to pons prevented the positive interhemispheric transfer effect.130,298 It also appears that the midline brainstem participates in symmetrical patterning and electroclinical tonic expression of the kindled seizure. It is possible that the kindling process removes inhibitory influences of specific (as yet unidentified) brainstem regions and that this removal of inhibition contributes to the progressive enhancement of limbic seizure susceptibility. Presumably, brainstem structures homolateral to the primary kindling site must participate in the progression of kindling at that site via intrahemispheric projections. This notion is supported by the observation that unilateral lesions of the midbrain reticular formation markedly reduce susceptibility to seizures evoked by kindling of the homolateral amygdala in cats.299 Apparently, the bisection of the brainstem can prevent the kindling-induced changes from occurring in the contralateral hemisphere, rendering that hemisphere “naive” to the kindling stimulation.
Kindling of Brainstem Regions
The anatomic separation between forebrain and hindbrain seizures that has been repeatedly documented in experimental models is largely derived from the study of acute seizures in otherwise normal animals. Under conditions in which repeated seizure activity or kindling has occurred over prolonged periods, an erosion of this anatomic separation may take place. For example, repeated audiogenic seizures (daily for 2–3 weeks) give rise to seizures that resemble forebrain seizures, based on behavioral and electrographic characteristics. Moreover, audiogenic kindling in susceptible animals markedly accelerated hippocampal and amygdala kindling.138,139 Data from 2-deoxyglucose, c-Fos protein expression, and functional disconnection by local lidocaine injections, indicated that the amygdala, but not hippocampus, is critical for the spread of audiogenic seizures from brainstem to forebrain.138
Similarly, whereas acute electrical stimulation of the inferior colliculus evokes seizures limited to the hindbrain, repeated electrical or acoustical stimulation of this structure eventually recruits forebrain circuits and induces forebrain seizure discharge.139,140,186 Amygdala spiking has been seen after chronic inferior colliculus stimulation, indicating forebrain recruitment.116,117 Once forebrain circuits have been recruited by repeated inferior colliculus stimulation, the focal inhibition of area tempestas (an epileptogenic region within the deep rostral piriform cortex) can suppress the limbic motor seizures that occur as a result of seizure spread into the forebrain. This suggests that the kindling-evoked spread of seizure discharge into the forebrain comes under the control of forebrain circuits that do not otherwise influence brainstem–evoked seizures.182
Other studies demonstrate that repeated seizures in forebrain circuits can modify susceptibility to brainstem convulsions. For example, amygdala kindling was found to increase susceptibility to electroshock-induced tonic hindlimb extension, even though the kindled seizures themselves never evoked a tonic extensor response.7 Apparently, repetitive seizure activity in one circuit may alter the susceptibility of other circuitry to seizure induction, possibly by inducing long-term changes in structures and pathways (e.g., substantia nigra, midline thalamic nuclei, superior colliculus, ascending noradrenergic projections) that serve a general gating function (see later discussion).
Seizure-Gating Mechanisms in Basal Ganglia and Brainstem
Under this category, we describe certain pathways that influence seizure susceptibility by modulating the threshold for seizure initiation or by regulating the sensitivity of the pathways responsible for propagating the seizure activity.
In many cases, the gating substrates are relatively nonselective as to the type of seizure they can influence. Consequently, by manipulating the activity of seizure gating systems, we can exert a broad-spectrum shift in seizure susceptibility. There are, however, circumstances in which a particular pathway exerts divergent or opposing effects on different types of seizures, further reinforcing the independent nature of some of the seizure-generating networks.
Basal Ganglia and Related Structures
The earliest indications that the basal ganglia participated in seizure propagation came from subcortical ablation stu-dies133,145 that identified the substantia nigra (SN) and the thalamic and lenticular nuclei as crucial for the generalization of chemically evoked cortical seizures. Depth recording experiments80 showed involvement of the putamen and associated structures in the spread of seizures evoked focally from cortex or amygdala; similarly, seizures evoked from hippocampal stimulation or by systemic chemoconvulsants were observed to propagate through the SN.134 Metabolic mapping studies reinforced the involvement of the basal ganglia in seizures by demonstrating a profound increase in 2-deoxyglucose accumulation in the SN and globus pallidus after kindled amygdala seizures.79 More recent studies have suggested that increases in metabolic activity in basal ganglia nuclei may precede the onset of clinical seizures during epileptogenesis: SN and striatum are hypermetabolic in rats that are at the end of the latent period following lithium-pilocarpine status epilepticus (SE); that is, at a time point just prior to the development of spontaneous seizures.71,72 Likewise in 21-day-old genetically susceptible rats (GAERS) that do not yet exhibit spike-and-wave discharges (SWDs), rates of glucose utilization are increased in the SN, superior colliculus, and globus pallidus.221 It is possible that these changes reflect ongoing subclinical seizure activity and/or an effort of the network to retard or prevent the occurrence of seizures.
The influence of the basal ganglia on seizure propagation and generalization is not confined to the motor manifestations of the seizures. In instances in which electrographic signs of seizures have been monitored, anticonvulsant manipulations of basal ganglia structures induce a corresponding suppression of electrographic seizure discharge in cortical and subcortical structures.112,162,192,271,272,275,276 This is consistent with a role of basal ganglia circuits in the control of cortical excitability and the regulation of neuronal discharge synchronization in widespread regions of the forebrain.
Substantia Nigra
Broad-Spectrum Seizure Protection
The caudate-putamen (striatum), globus pallidus (entopeduncular nucleus in particular), and SN are all basal ganglia components that have been demonstrated to influence seizure susceptibility in numerous experimental seizure models. The best studied of these structures is the SN, the only region that has been examined in connection with more than 10 different experimental seizure models.100,101
Bilateral treatments that either increase inhibitory transmission (mediated by the neurotransmitter GABA) in SN, or block excitatory transmission (mediated by glutamate or certain neuropeptides such as substance P) in the SN pars reticularis (SNpr) prevent or attenuate convulsive seizures induced by several chemoconvulsants (pilocarpine, kainic acid, bicuculline, and flurothyl, among others), maximal electroshock, kindling of amygdala, drug application into the area tempestas, acoustic stimulation in rats susceptible to audiogenic seizures (the subject of several reviews60,100,101,105,284,289), and spontaneously occurring in absence-like seizures in GAERS.65 These anticonvulsant effects may be mediated largely by the rostral regions of the SNpr, because activation of GABA receptors in the caudal SNpr has been reported to be proconvulsant in the flurothyl model.214,268,291 Velíšková and Moshé have reviewed regional differences within the SNpr.289
Not only motor manifestations, but also electrographic manifestations of seizures are regulated by SN: Inhibition within the SNpr decreases susceptibility to nonconvulsive electrographic SWD that occur either spontaneously (in genetically predisposed strains of animals such as GAERS) or as a consequence of treatment with drugs such as pentylenetetrazol in low doses.58,60,61,62 Although the GABA agonist muscimol in SNpr was found not to protect against seizures evoked by high doses of pentylenetetrazol315 blockade of glutamate transmission in SN did protect against these seizures,310 indicating a role for SNpr in the modulation of seizures in this model. The lack of anticonvulsant effects of intranigral muscimol found in the latter study may be due to the high doses of pentylenetetrazol employed; these doses are likely to interfere with GABA receptor function and the postsynaptic responses to muscimol in the SNpr. In fact, seizures induced by lower doses of pentylenetetrazol are attenuated by SNpr muscimol infusions.144,222 Other pharmacologic manipulations of SNpr that are anticonvulsant include augmenting serotonergic transmission,226 stimulating opiate receptors,110 blocking substance P receptors,111 and enhancing adenosine transmission.136 The anticonvulsant effect of adenosine and the proconvulsant effect of adenosine antagonists such as caffeine and theophylline within the SN suggest that the synaptic availability of adenosine may provide a physiologic mechanism whereby seizures can be self-limiting.136
The SNpr is probably part of a seizure-suppressing circuit that becomes engaged by seizure discharge. For example, the SNpr of kindled animals becomes recruited into a burst-firing pattern during the afterdischarge.21 Increased burst firing in SNpr can still be seen 1 day after a fully kindled seizure, with a significant increase in neuronal discharge rate in the posterior, but not anterior, SNpr.120 Likewise, in GAERS, simultaneously with the occurrence of SWDs at the cortical level, SNpr neurons increase their firing rates, and action potentials become organized in bursts. However, this SN burst-firing pattern stops before or at the end of the nonconvulsive seizure.62,63 Thus, the SN and associated nuclei of the basal ganglia may act to maintain a homeostatic balance of brain excitability, attempting to resist the development of widespread synchrony.
The SN also may be a substrate for the actions of several antiepileptic drugs. Bilateral injections of midazolam, phenobarbital, or trimethadione into the SN protect against systemic pilocarpine seizures in a rodent model. Not surprisingly, ethosuximide had no effect,270 because the site of action of ethosuximide has been localized to the hindbrain.176
The integrity of the SN is not required for seizure induction, indicating that this structure is not part of a crucial seizure-conducting pathway. Bilateral destruction of the SN has been shown by Garant and Gale109 and by McNamara et al.192 to protect against experimentally evoked seizures by shifting the threshold for seizure induction. Although a later study301 reported no effect of bilateral destruction of SN on kindled seizures (in direct contrast to the findings of McNamara et al.), the completeness of nigra damage in that study is uncertain in the absence of histologic documentation of the lesions.
Role in Genetic Predisposition to Epilepsy
The SN may be a site at which genetic abnormalities can determine seizure susceptibility. For example, it has been suggested that abnormal GABA mechanisms in the SN may contribute to the seizure susceptibility in the GEPR. Clobazam, a benzodiazepine, focally administered in the SN blocked audiogenic seizures in normal rats made susceptible to these seizures by administration of bicuculline in the inferior colliculus. However, intranigral clobazam did not block audiogenic seizures in the GEPR. Because clobazam acts by enhancing endogenous GABA transmission, this indicates that endogenous GABA transmission in the nigra of GEPRs may be deficient.266 Moreover, intranigral injections of muscimol had no effect on audiogenic seizures in a seizure-prone strain of Wistar rat.57 Franck and Schwartzkroin92 found a defect in GABA receptor binding in the SN of GEPRs but did not find it in the inferior colliculus. Microdialysis studies have shown that potassium (K+)-evoked GABA release within the SN is significantly reduced in the GEPR, as compared with controls.69
Role of GABA, Glutamate, and Other Neurotransmitters
The fact that GABA agonists in the SNpr reduce seizure susceptibility in seizure models in which depletion or blockade of GABA in the SNpr does not increase seizure susceptibility,172,225 suggests that GABA release is not the only mechanism by which seizure resistance can be achieved from the SN. Enhancing 5HT transmission in SN also confers seizure resistance.224,226 GABA and 5HT appear to work in concert with each other in the SN: Under conditions in which GABA transmission is impaired, 5HT release can increase to maintain inhibitory tone.99 Consequently, when 5HT is depleted, the blockade of GABA receptors in the SNpr becomes proconvulsant.225 Therefore, a defect in 5HT transmission may compromise the endogenous anticonvulsant mechanisms in the SN and predispose to epilepsy.
Consistent with the role of GABA in the SN serving a seizure gating function and not a seizure triggering function is the observation that seizures are not triggered by the blockade of GABA receptors in the SNpr,172 even in the presence of 5HT depletion.
The only intranigral treatment that has been documented to trigger clinical seizures is kainic acid (KA). The presence of KA focally in the SN can provoke a long duration state of SE in the Sprague-Dawley rat.171 The fact that the seizures occur following a considerable latency suggests that the KA-induced stimulation of a subpopulation of SNpr outputs is sufficient to lower seizure threshold in limbic networks to the extent that SE can develop from spontaneous (endogenous) patterns of activity in limbic circuits. Because intranigral GABA agonists are ineffective in blocking the development of seizures following intranigral KA, it appears that the kainate stimulation
overwhelms GABAergic control in this region.171 This may explain why SE induced by systemic KA is resistant to the anticonvulsant action of intranigral GABA agonists.
overwhelms GABAergic control in this region.171 This may explain why SE induced by systemic KA is resistant to the anticonvulsant action of intranigral GABA agonists.
Sources of Afferent Control
The activity of SN neurons can be affected by epileptogenic influences through several routes. The major route is via the striatum, which receives information converging from widespread cortical regions and communicates to the SN via the “direct” striatonigral projections, which are largely GABAergic and inhibitory on nigral neurons. In addition, cortico-subthalamic-nigral pathways relay cortical influences to the SN; these tend to be excitatory on SNpr neurons.179 At the same time, ascending 5HT and noradrenergic projections from the brainstem modulate nigral activity in response to changes in arousal, pain, sleep stages, and associated fluctuations in autonomic and endocrine function.
Stimulation of various cortical regions evokes predominan-tly inhibitory responses in the SNpr, most likely reflecting transmission mediated via the cortico-striatal-nigral pathways.152 The cells in the SNpr that respond to stimulation of the prefrontal cortex are distinct from those responsive to auditory cortex stimulation, and these populations are differentially localized in SNpr, suggesting a topographic organization and functional compartmentation within the SNpr circuitry.152
Striatum
Certain key structures of the forebrain and midbrain work in concert with the SN in a seizure-resisting capacity (see Fig. 1). The striatum is a major source of neural input to the SN, whereas the superior colliculus (deep layers) is an important target of neural projections coming from the nigra. One of the most prominent pathways connecting the striatum to SN is inhibitory and utilizes GABA as its transmitter.91,132 Likewise, the SN sends an inhibitory GABA-containing projection to the superior colliculus.67,296 Consequently, it might be expected that the stimulation of the neurons in the striatum that give rise to the GABA inputs to the SN would enhance GABA transmission in the SN and thereby exert an anticonvulsant effect. The experimental evidence supports this proposal: Electrical or drug-induced excitatory stimulation in striatum is anticonvulsant in experimental seizure models.5,34,159,276 LaGrutta et al.159 found that a prior conditioning stimulation to the caudate nucleus (30 c/s, 1.5 msec for 5 s, 5 V) caused almost complete inhibition of the afterdischarge in amygdala, hippocampus, and temporal cortex evoked by stimulation of a kindled amygdala.
The anticonvulsant effect of GABA blockade in striatum can be reversed by blocking GABA in the SN or entopeduncular nucleus (see discussion in next section), consistent with the concept that GABAergic efferents from striatum mediate this region’s influence on seizure susceptibility.
Entopeduncular Nucleus (Globus Pallidus, Internal Segment)
The entopeduncular nucleus, also known as the internal or medial segment of the globus pallidus, is similar to the SN in function, morphology, and neuroanatomic connections. Together with the SN, this nucleus relays neuronal outputs from the striatum. It is therefore not surprising that the same treatments that suppress seizure propagation when placed in the SN exert a similar action in the entopeduncular nucleus.51,229 However, unlike the SN, the entopeduncular nucleus does not appear to exert an influence on brainstem seizure substrates. Whereas blockade of glutamate transmission in SN attenuated audiogenic seizures in the GEPR, this manipulation in entopeduncular nucleus did not reduce audiogenic seizure activity193 under the same conditions in which it blocked pilocarpine-induced limbic motor seizures.229 Moreover, GABA agonist application in entopeduncular nucleus did not prevent maximal electroshock-induced tonic convulsions under conditions in which it prevented pilocarpine-induced limbic motor seizures.141 Thus it appears that the influence of the entopeduncular nucleus may be preferentially directed at forebrain seizure circuitry, perhaps via its close connections with the habenula and other limbic structures.
Globus Pallidus: External Segment (GPe)
The indirect pathway of the basal ganglia system is a polysynaptic circuit composed of (a) GABAergic striato-pallidal neurons projecting to the external segment of the globus pallidus, (b) GABAergic neurons projecting from GPe to the subthalamic nucleus (STN)4 and to SN, and (c) neurons projecting from the STN to SN.4 Because the GPe sends GABAergic projections into SN, it is expected that the activation of these projections would attenuate seizures. This has shown to be the case in the GAERS model, where the disinhibition of GPe by local injection of the GABAA antagonist, picrotoxin, at doses devoid of behavioral effects, suppressed nonconvulsive absence seizures.66 The antiepileptic effect obtained at the pallidal level is also linked to a decrease in glutamate levels in the rat SN measured by microdialysis,66 suggesting that a reduction of activity in STN may also contribute to the seizure attenuation (see discussion in the next section).66 On the other hand, disinhibition of GPe did not attenuate amygdala kindled seizures,62 indicating that the role of this structure in seizure control may be more limited than the role of the SN. Interestingly, repeated stimulation of the GPe is effective for generating kindled seizures, suggesting that seizure suppressing actions of acute GPe stimulation are easily overridden in chronic conditions.
In sharp contrast to these results, bilateral GABAergic inhibition within the GPe, using the GABA uptake inhibitor tiagabine or a GABAB agonist, baclofen, markedly attenuated the tonic convulsions induced by acute pentylenetetrazol treatment.39 This observation raises the possibility that GABAB receptors may exert control on a subpopulation of GPe projections that are distinct from those regulated by GABAA receptors. Thus, the role of GABAergic synapses in GPe on the development of seizures is likely to be varied and highly dependent on the nature of the seizure. The fact that GABAergic synaptic transmission in the GPe is especially sensitive to rapid frequency-dependent depression238 may account for the conditional nature of the influence of the GPe on seizure activity.
Subthalamic Nucleus
Interactions with the Substantia Nigra
As an important component of the basal ganglia network, the STN represents a source of input into both the SN and the entopeduncular nucleus (GPi). It is therefore well positioned to regulate the outflow from these critical nuclei. Direct afferents to the STN arise from the prefrontal, premotor, and motor cortex; these projections are largely excitatory, mediated by glutamate. Inhibitory projections to STN arise form GPe; these relay the indirect influence of the cortico-striatal system.
Detailed analysis of the nature of STN interactions with the SN have largely been confined to the rat. In the rat, the STN provides a major source of glutamatergic excitatory drive that sustains the firing of nigral neurons.247,254,256 When
activity in the rat STN is inhibited either by GABA agonists or glutamate antagonists, this results in a decreased activity of SNpr neurons.87,247 Thus, it stands to reason that lesions or inhibition of STN could mimic the anticonvulsant action of inhibition in the SNpr. Experimental evidence in support of this proposal derives from the studies of Deransart et al. in amygdala kindled rats or GAERS,64 Dybdal and Gale77 in rats with seizures evoked focally or systemically with bicuculline, and Velíšková et al.290 in rats with flurothyl-induced seizures. As in the case of SNpr inhibition, bilateral STN inhibition is required for anticonvulsant effects.64,77,290
activity in the rat STN is inhibited either by GABA agonists or glutamate antagonists, this results in a decreased activity of SNpr neurons.87,247 Thus, it stands to reason that lesions or inhibition of STN could mimic the anticonvulsant action of inhibition in the SNpr. Experimental evidence in support of this proposal derives from the studies of Deransart et al. in amygdala kindled rats or GAERS,64 Dybdal and Gale77 in rats with seizures evoked focally or systemically with bicuculline, and Velíšková et al.290 in rats with flurothyl-induced seizures. As in the case of SNpr inhibition, bilateral STN inhibition is required for anticonvulsant effects.64,77,290
A similar profile of anticonvulsant effects has been achieved in the rat using high-frequency (130 Hz) electrical stimulation of the STN.161,277,292 This effect is frequency-dependent,161 and the frequency-dependency is closely related to the ability of the stimulation to cause a decrease in the activity of STN neurons263 as well as SN neurons.180,263 Although “depolarization block” has been proposed to account for the inhibitory action of the high-frequency stimulation of STN, the fact that the inhibitory action in STN is rarely complete263 and that it is reversed by iontophoretic application of a GABA receptor antagonist180 argues against this proposal. Instead, it appears that the stimulation in STN may activate fibers that cause the release of GABA into the SNpr.180,307,308 These fibers could be striatonigral, pallidonigral, and/or collaterals of the SNpr GABAergic neurons themselves.
Interactions with the Frontal Cortex
An effect on SNpr may not be the only mechanism by which STN stimulation in the rat can suppress seizures. Usui et al.277 found that unilateral stimulation of STN was effective in preventing the recruitment of frontal cortex into the electroencephalographic seizure discharge induced by systemic KA in rats, even though this treatment was ineffective in reducing the total seizure duration or the incidence of seizure discharge recorded from hippocampus (which was actually increased by the STN stimulation). This contrasts sharply with the more robust anticonvulsant effects observed with bilateral STN stimulation.161,292 Because inhibition of SN must be bilateral to achieve an anticonvulsant action, it is likely that the unilateral stimulation of STN influences the seizure discharge in the frontal cortex through a mechanism not mediated by SN. In fact, in the same study, Usui et al.277 found unilateral high-frequency stimulation in the SNpr to be ineffective, in agreement with previous observations281 and in contrast to the anticonvulsant actions of bilateral stimulation in the SNpr.281
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


