7 Note: Significant diseases are indicated in bold and syndromes in italics. Types of Memory by Subject Declarative/episodic—knowledge of events (hippocampus) Procedural—riding a bike (basal ganglia, cerebellum) Conditioning—associative learning (cerebellum) Semantic—factual or definitional knowledge (hippocampus-independent) Types of Memory by Duration Immediate/working (seconds) Short-term/anterograde (< 10 minutes) Long-term/remote (> 10 minutes) 1. Definition: cognitive impairment that does not achieve the criteria for dementia because the degree of impairment does not significantly affect the patient’s activities of daily living 2. Subtypes a. amnestic MCI/age-related memory loss: an objective impairment of memory function in comparison with the normal range for the patient’s age, which does not involve significant impairment of other cognitive functions or impair the patient’s activities of daily living i. 15% of cases/year develop into Alzheimer’s disease, which is ~ 10 times the incidence of non-MCI patients; the incidence of progression to Alzheimer’s disease is increased in MCI patients who express the apolipoprotein E (APOE) ε4 allele (1) small hippocampal and/or entorhinal cortex volumes on a MRI scan may also predict progression of MCI to Alzheimer’s disease, although the rate of volume loss is not helpful b. MCI with impairment in a nonmemory domain (Box 7.2) c. MCI with impairment in multiple domains 3. Treatment: none yet established 1. In general, impairment of at least two of five functional domains (memory, language, visuospatial, emotion, executive) that affects the patient’s activities of daily living a. dementia is usually suspected in patients who have a mini-mental status exam (MMSE) score < 24, but performance is dependent upon the patient’s age and education background; therefore, use the MMSE only for screening, not for establishing the diagnosis i. MMSE < 24 often does not identify non-Alzheimer’s dementias 2. Subtypes of dementia: cortical versus subcortical dementias (Table 7–1) 1. Reversible causes exist in 10% of dementia cases, and include a. metabolic abnormalities: hyponatremia, hypocalcemia, hypoglycemia b. organ-failure: respiratory failure (hypoxia or hypercarbia); liver, renal, or cardiac failure
Behavioral Neurology and Psychiatry
I. Dementias
A. Mild Cognitive Impairment (MCI) (Box 7.1)
Box 7.1
B. Definition of Dementia
C. Reversible Causes of Dementia
Functional domain | Cortical dementia | Subcortical dementia |
Memory | Learning > retrieval deficit | Retrieval > learning deficit |
Language | Aphasic | Dysarthric |
Emotion | Disinhibited | Depressed |
Visuospatial | (Indistinguishable) |
|
Executive | Impaired and does not improve with cues | Impaired, but improves with cues |
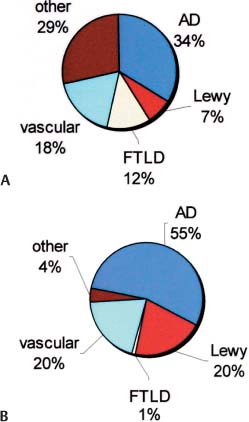
Figure 7–1 Epidemiology of major dementing disorders. (A) <65 years old; (B) >65 years old. (AD, Alzheimer’s dementia; Lewy, Lewy body dementia)
c. endocrine disorders: hypo- and hyperthyroidism, hyperparathyroidism, adrenal failure or Cushing’s syndrome
d. toxin-induced: heavy metal exposure, drugs, alcohol
e. vitamin deficiency, particularly B1/thiamine or B12
f. medication use, particularly benzodiazepines, opioids, and anticholinergic agents
g. chronic brain infections (e.g., HIV, syphilis) or inflammatory conditions
h. intracranial mass lesions or hydrocephalus
i. normal pressure hydrocephalus
D. Epidemiology of Major Dementing Disorders (Fig. 7–1)
E. Alzheimer’s Dementia (AD)
1. Histology: primary cortical atrophy involving the loss of synapses, and neurons, and the shrinkage of surviving neurons; atrophy initiates in the mesial temporal and posterior cingulate cortex (cortex layers V-VI) (Box 7.3), but eventually involves the hippocampus and all of the cortex sparing only the primary motor, sensory, and visual cortices
Box 7.3
Cortical histopathology in AD is in comparison to the loss of layers I–III in frontotemporal lobar degeneration disorders
a. neurons in the nucleus basalis of Meynert, locus coeruleus, and raphe are also reduced in number early in the disease
b. hippocampal atrophy is predominant in the CA1 area (unlike Lewy body dementia, which affects CA2–3)
c. the cortex and hippocampus lose choline acetyltransferase > nicotinic and muscarinic acetylcholine receptors well into the course of the disease, therefore these are not the primary defects; also exhibit reduced acetylcholine release and decreased choline reuptake (necessary for acetylcholine synthesis), which may relate to a decreased trophic responsiveness to nerve growth factor
d. plaques and tangles (Fig. 7–2)
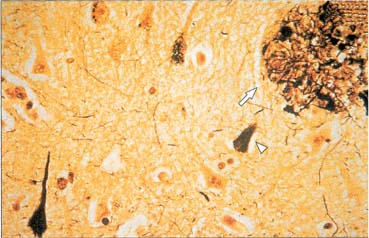
Figure 7–2 Neurofibrillary tangles (arrowhead) and neuritic plaque (arrow) in Alzheimer’s disease. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:123, Fig. 121. Reprinted by permission.)
i. neurofibrillary tangle: intraneuronal inclusions of hyperphosphorylated tau protein (a microtubule-associated protein) and ubiquitin (which binds and marks other proteins for degradation)
(1) neurofibrillary tangles are associated with an early age of disease onset and a more severe disease course
(2) neurofibrillary tangles may not occupy the whole neuron, and may only fill a dendrite {neuropil thread}
ii. plaques: all are extracellular
(1) neuritic/senile plaque: composed of abnormal neuronal processes (dystrophic neurites), and glial processes surrounding an amyloid core; also includes tau and other molecules (acetylcholinesterase, ubiquitin, chymotrypsin, complement proteins, glycosaminoglycans)
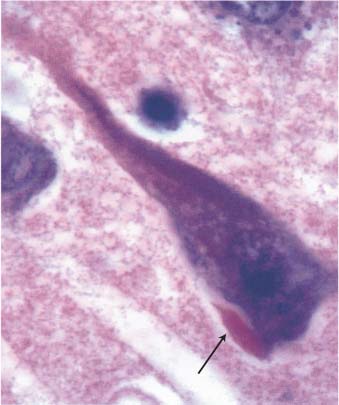
Figure 7–3 The Hirano body (arrow). (From Hirano A. Color Atlas of Pathology of the Nervous System, 2nd Ed. Tokyo/New York: Igaku-Shoin Press; 1988:94, Fig. 222. Reprinted by permission.)
(2) ghost plaque: develop only after cell death when extracellular proteases degrade the neurofibrillary tangle, leaving only the dense amyloid core
(3) diffuse plaque: similar histological appearance to senile plaques; composed of amorphous nonfibrillar β-amyloid or finely fibrillar β-amyloid deposits
iii. Hirano bodies (Fig. 7–3): eosinophilic intracellular inclusions in hippocampal pyramidal cells that have a “herringbone” appearance on electron microscopy
iv. Lewy bodies (see p. 162): more commonly found in the synucleinopathies (Parkinson’s disease, Lewy body dementia, multisystem atrophy), but are present in the substantia nigra in 20% of AD
e. amyloid angiopathy caused by the β-amyloid protein coexists in 90% of AD patients (see p. 65)
2. Subtypes
a. sporadic AD (95% of cases): symptomatic onset > 65 years of age, typically with a slowly progressive course at time of diagnosis (i.e., at the time of diagnosis, the patient is 2–3 years into the course of the disease)
i. risk factors
(1) increasing age
(2) APOE alleles: APOE is component of very low-density lipoprotein (VLDL) and chylomicron particles, which facilitates their uptake by muscle and fat cells (Box 7.4)
Box 7.4
Other apolipoprotein disorders–APOA1 deposits in some types of amyloidosis; APOB deficiency in abetalipoproteinemia
(a) the APOE ε4 allele is present in 50% of sporadic AD cases; no clear association with the ε4 allele and the severity of the disease
(b) APOE ε4 binds β-amyloid, creating an insoluble complex that may form plaques
(c) APOE ε3 allele may stabilize tau protein thereby preventing neurofibrillary tangle formation, but only the rare APOE ε2 allele is known to be protective against AD
(3) family history of AD without clear inheritance pattern
(4) low level of education
(5) female gender
(6) history of severe head trauma
(7) stroke and vascular risk factors
(8) small head size
(9) Down’s syndrome
b. familial/early-onset AD (5% of cases): patients are generally < 65 years old at onset, and exhibit a more rapid progression of symptoms than do sporadic AD patients; although the disease course is still insidious
i. causative genetic mutations (Fig. 7–4): all have autosomal dominant inheritance
(1) the AD1 mutation in the amyloid precursor protein (APP) gene (chromosome 21), which occurs in a region that codes for the β-amyloid protein, accounts for 2% of familial AD
(a) overexpression of the APP in trisomy 21/Down’s syndrome may account for the early-onset AD in these patients
(2) presenilin 1 (PS1, chromosome 14): accounts for 75% of familial AD (Box 7.5)
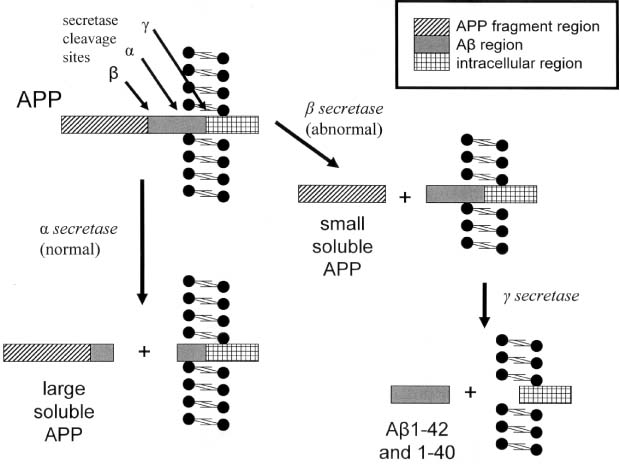
Figure 7–4 Key enzymes of Alzheimer’s disease. (APP, amyloid precursor protein)
(3) presenilin 2 (PS2, chromosome 1)
(4) 2 macroglobulin (chromosome 12)
3. Epidemiology
a. age distribution: prevalence at 60 years of age = 3%; prevalence at 90 years of age = 50%
b. prevalence as a cause of dementia is likely underestimated because
i. Lewy body dementia has a high coincidence with AD
ii. ~ 50% of vascular dementia cases have elements of AD histology on postmortem examination
4. Symptoms: course of impairment is typically memory loss before involvement of any other cognitive functions or behavioral impairment
a. memory loss: early and severe impairment of short-term memory > long-term or immediate memory
i. episodic memory is most severely affected
ii. procedural memory is well-preserved, although mild apraxia can be detected early in the disease
b. language impairment: initial language abnormality is anomia (due to lateral temporal dysfunction, not memory impairment) that progresses to a transcortical sensory aphasia
c. behavioral impairment: more common late in the disease
i. agitation and aggressive behaviors (70%), but rarely sexual disinhibition (10%)
ii. depression (50%), which is proportionate to cell loss in locus coeruleus
iii. simple delusions (30%) that are often paranoid in nature
iv. hallucinations (20%)
d. visuospatial impairment: manifests primarily as difficulty with navigation
e. executive impairment: impaired judgment and reasoning, and poor abstraction and decision-making abilities occur late in the disease as frontal lobes become involved (Box 7.6)
Box 7.6
Agnosias can occur in AD, but they are distributed topographically (e.g., visual agnosias to the occipital lobe).
f. generally does not involve focal neurological signs, seizures, motor disturbances (i.e., parkinsonism), or impairment in the level of arousal until late in the disease; the physical exam generally is nonfocal, although multiple frontal lobe release reflexes (e.g., glabellar reflex, grasp, snout reflex) are often present (Box 7.7)
Box 7.7
20% of normal young persons have one frontal lobe release sign, but the palmomental reflex is always pathological
i. myoclonus occurs in 10% of AD cases, which can be confused with prion diseases
ii. pyramidal signs (increased reflexes, Babinski reflexes, hypertonus) are unlikely to be due to AD, as the primary motor and sensory cortices are not involved in the disease process until very late
5. Diagnostic testing: none are well-established or necessary for diagnosis
a. genetic markers: the low incidence of familial AD makes them impractical as screening tools
b. cerebrospinal fluid protein markers: protein markers correlated with pathologically determined AD include
i. increased tau protein and hyperphosphorylated tau protein levels
ii. combined increases in β-amyloid protein and tau protein levels
c. EEG: loss of alpha activity and increased theta and delta frequencies in 80% of AD cases with > 4-year disease duration; useful for differentiating against the frontotemporal lobar degeneration disorders in which EEG is relatively normal
d. neuroimaging
i. SPECT scans demonstrate hypoperfusion in the temporal and parietal lobes
ii. PET scans demonstrate hypometabolism in the temporal and parietal lobes
iii. MRI and CT demonstrates reduced hippocampal, entorhinal cortex, and posterior cingulate cortex volumes (Fig. 7–5)
6. Treatment
a. preventative
i. chronic use of NSAIDs is associated with a decreased risk of developing AD, but it is not an established prophylactic treatment
ii. hormone replacement therapy appears to increase, not reduce, the risk of dementia
b. symptomatic treatment
i. acetylcholinesterase inhibitors: donepezil (Aricept), rivastigmine (Exelon), galantamine (Reminyl)
(1) improve functional abilities and delay institutionalization in patients with mild or moderate dementia; also appear to be beneficial in severe dementia
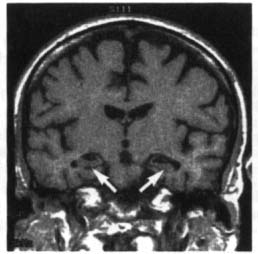
Figure 7–5 Coronal T1 MRI from a patient with Alzheimer’s disease. Note the pronounced hippocampal atrophy (arrows). (From Nestor P, Hodges J. Non-Alzheimer’s dementias. Semin Neurol 2000, 20:443, Fig. 1b. Reprinted by permission.)
(2) tacrine also improves cognitive performance but it has a high rate of side effects (particularly hepatotoxicity) and requires q.i.d. dosing, therefore it is rarely used
ii. memantine: acts as an N-methyl-D-aspartate (NMDA) glutamate receptor antagonist; improves patient’s functional abilities and reduces caregiver burden in patients with moderate or severe dementia
iii. no evidence for use of vitamin E, NSAIDs, or estrogens
c. treatment of behavioral impairment
i. depression: SSRIs or trazodone, which have minimal anticholinergic side effects
ii. agitation: behavioral modification; buspirone; atypical antipsychotics (olanzapine, quetiapine, risperidone, clozapine)
(1) avoid benzodiazepines because they worsen cognitive function
iii. insomnia: improve sleep hygiene, avoid daytime naps; trazodone
Type | Symptoms different from AD | Signs different from AD |
Pseudodementia/depression with cognitive disturbances | Rapid onset | Good performance on cognitive tests; self-referral to physician |
Vascular dementia | Classically (but not commonly) has an abrupt onset and stepwise deterioration | Focal deficits; bulbar signs |
Lewy body dementia | Fluctuating cognitive abilities & attention; visual hallucinations; sleep disturbance; frequent falls from syncope | Parkinsonism; high sensitivity to the side effects of antipsychotics |
Frontotemporal lobar degeneration/Pick’s disease | Impulsivity; compulsivity; emotional lability; disinhibition; aphasia | Lack of EEG slowing; lack of visuospatial impairment or other parietal lobe findings |
Prion dementias (e.g., Creutzfeldt-Jakob disease) | Abrupt onset, rapid progression; early aphasia; extrapyramidal symptoms | Cerebellar symptoms; visual loss; myoclonus; CSF positive for 14–3-3 protein; diffuse slow-wave EEG with periodic complexes; neuroimaging abnormalities (see p. 265) |
Parkinson’s disease | Subcortical dementia features; subcortical dementia symptoms | Parkinsonism |
Huntington’s disease | Impulsivity; hypersexuality; psychosis; subcortical dementia symptoms | Chorea |
Abbreviations: CSF, cerebrospinal fluid; EEG, electroencephalogram.
7. Prognosis
a. nursing home placement occurs on average 3 years after diagnosis
b. 70% 5-year mortality, with death usually being attributed to complications of immobility or malnutrition (Table 7–2)
F. Vascular/Multi-Infarct Dementia
1. Pathophysiology: caused by any combination of cortical infarction, lacunar infarction, and/or leukoaraiosis; also involves microscopic areas of neuron loss and gliosis {microinfarcts}
a. requires ~ 50 cc of tissue loss to achieve symptomatic dementia
b. risk factors are as per stroke, but also include the cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) gene mutation and the APOE ε4 allele (as in AD)
c. epidemiology: incidence increases with age; 25% of patients will become demented within 3 months of a stroke
2. Subtypes
a. large infarct dementia: caused by a few cortical and/or subcortical infarcts that are larger than lacunes
b. small infarct dementia: typically caused by multiple lacunes {l’etat lacunaire}, leukoaraiosis, CADASIL, or amyloid angiopathy
c. strategically placed infarct dementia: caused by single lesions in the hippocampus/medial temporal lobe, medial thalamus, caudate nucleus, or nondominant parietal lobe; such infarcts tend to have a preference for the dominant hemisphere
d. hypoxic-ischemic encephalopathy
e. hemorrhagic dementia secondary to subdural hemorrhage, subarachnoid hemorrhage, or intracerebral hemorrhage
4. Diagnostic testing
a. the size and exact number of infarcts are not as predictive of dementia as is cortical atrophy
b. isolated lacunar infarcts occur in 20% of the general population, but are associated with worse cognitive performance and an increased risk of dementia
c. periventricular white matter T2 hyperintensities on an MRI scan {leukoaraiosis} is not specific for vascular dementia or any dementia, but is associated with worse performance on cognitive tests
5. Treatment: as per stroke prevention, with particular attention to the reduction of hypertension (reduces incidence of dementia by 50%) and reduction of elevated homocysteine levels; acetylcholinesterase inhibitors; memantine
6. Prognosis: 30% 5-year survival, which is worse than other types of dementia or nondemented stroke; however, cognitive deterioration progresses more slowly than in AD
G. Frontotemporal Lobar Degeneration/Pick’s Disease (Box 7.8)
Box 7.8
Frontotemporal lobar degeneration is not the same as frontotemporal dementia, which is a subtype of it
1. Histology: Selective atrophy of the frontal (particularly orbitofrontal) and/or anterior temporal lobes often involving the hippocampus, amygdala, and/or underlying basal ganglia (particularly the caudate); atrophy tends to spare the sensory cortices (as with AD) but also the parietal lobe (unlike AD)
a. pronounced astrocyte gliosis in cortex and subcortical white matter with loss of cortical layers I–III (versus AD that affects layers V–VI predominantly)
b. loss of acetylcholine receptors > choline acetyltransferase (in comparison with AD)
c. inclusion bodies: argentophilic cellular inclusions {Pick bodies} and swollen neurons {Pick cells} are fairly specific for the frontotemporal lobar degeneration disorders, although they are found in < 50% of cases (Fig. 7–6); cases with Pick bodies and cells tend to be more rapidly progressive
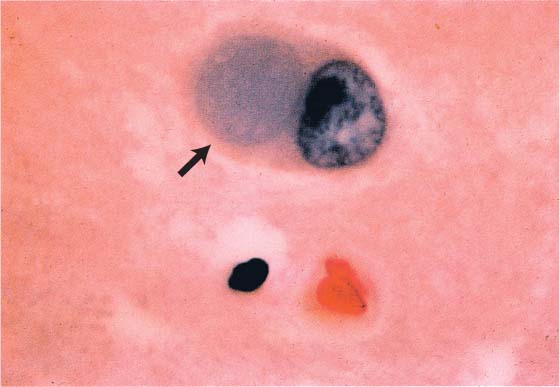
Figure 7–6 The Pick body (arrow). (From Hirano A. Color Atlas of Pathology of the Nervous System, 2nd Ed. Tokyo/New York: Igaku-Shoin Press; 1988:98, Fig. 233. Reprinted by permission.)
i. Pick bodies contain ubiquitin and tau protein tau
ii. in cases without Pick bodies or cells, intranuclear ubiquitin inclusions can often be identified
2. Pathophysiology
a. genetics: 20% of cases have a familial pattern that is usually autonomic dominant
i. tau gene mutations: mutations in the tau gene on chromosome 17 are responsible for 10% of familial frontotemporal lobar degeneration disorders, particularly those that involve parkinsonian features (i.e., the FTDP-17 variant)
ii. tau gene haplotypes: of the two tau haplotypes (H1 and H2) (Box 7.9), the H2 haplotype is particularly common in the semantic subtype of frontotemporal lobar degeneration disorders
Box 7.9
H1 haplotype is common in progressive supranuclear palsy and corticobasal ganglionic degeneration
b. associated with Parkinson’s disease and amyotrophic lateral sclerosis (ALS)
3. Subtypes: all have an insidious progression and symptomatic onset before 65 years of age (Box 7.10); lack of parietal lobe dysfunction differentiates against AD
Box 7.10
Eventually all frontotemporal lobar degeneration subtypes will progress to frontotemporal dementia.
a. frontotemporal dementia—common symptoms include impersistence of attention, apathy or disinhibition, and irritability; occasionally may also involve
i. Kluver-Bucy syndrome (usually incomplete): placidity, hypersexuality, hyperorality, and/or increased manual exploration
ii. perseverative behaviors with stereotypies
iii. echolalia and perseveration of speech progressing to mutism
b. primary progressive aphasia—caused by disease predominantly in the dominant frontal or temporal lobes
i. symptoms: aphasia of either a nonfluent or fluent type; progresses to dyslexia, agraphia, and apraxias, suggesting parietal lobe involvement
(1) isolated aphasia is usually present > 2 years before the development of other symptoms
c. semantic dementia—symptoms include reduced verbal fluency and pronounced naming difficulties; usually seen in patients with anterior temporal lobe atrophy
d. progressive prosopagnosia—usually seen in patients with nondominant temporal lobe (i.e., fusiform gyrus) atrophy
4. Diagnostic testing
a. neuroimaging may reveal an asymmetric atrophy pattern (Fig. 7–7)
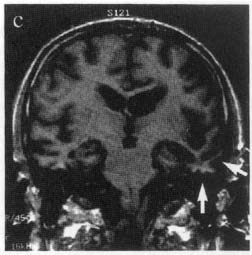
Figure 7–7 Coronal T1 MRI from a patient with semantic dementia. Note the pronounced atrophy of the left temporal lobe (arrows). (From Nestor P, Hodges J. NonAlzheimer’s dementias. Semin Neurol 2000, 20:443, Fig. 1c. Reprinted by permission.)
b. PET demonstrates hypometabolism and SPECT demonstrates hypoperfusion in the frontal and temporal lobes and often the basal ganglia
5. Treatment: None with proven efficacy; SSRIs for management of behavioral abnormalities irrespective of depression
H. Lewy Body Dementia
1. Histology: degeneration of the temporal and parietal cortices (like AD), but also of the occipital cortex; hippocampal atrophy predominantly affects the CA2–3 regions, unlike AD that predominantly affects CA1
a. exhibit significant loss of acetylcholine but limited loss of neurons in the cholinergic basal forebrain nuclei (i.e., basal nucleus of Meynert), unlike AD
b. exhibits loss of basal ganglia dopamine as in Parkinson’s disease, but also loss of basal ganglia D2 receptors
c. 80% of cases exhibit some Alzheimer’s-like histological abnormalities, particularly neuritic-like plaques that do not contain tau protein
d. inclusion bodies
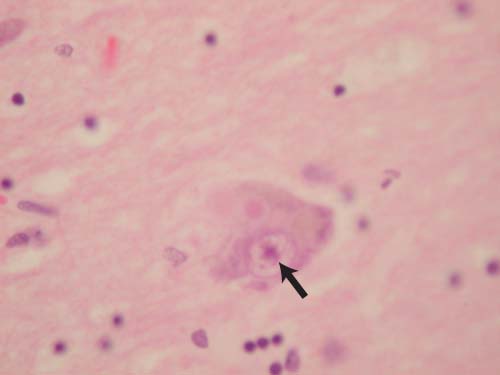
Figure 7–8 The Lewy body (arrow). Courtesy of Dr. C. Yamada.
i. Lewy body (Fig. 7–8): eosinophilic inclusions surrounded by a halo that contain α-synuclein and ubiquitin; found predominantly in the hippocampus and parahippocampal gyrus, but are widely distributed throughout the cortex, brainstem, and spinal cord (Box 7.11) (1) Lewy bodies are identical to those found in Parkinson’s disease
Box 7.11
Lewy body-like inclusions occur in Parkinson’s disease, Alzheimer’s disease, multiple system atrophy, corticobasal ganglionic degeneration, Hallervorden-Spatz disease, ataxia telangiectasia, and frontotemporal lobar degeneration ± amyotrophic lateral sclerosis.
ii. Lewy neurites: intracellular inclusions of ubiquitin and α-synuclein, which are limited to dendrites
2. Pathophysiology: Rare familial disease patterns are weakly associated with cytochrome P450 mutations or α-synuclein mutations
3. Symptoms
a. fluctuating cognitive abilities and level of arousal that may relate to cyclic sleep disturbance
b. autonomic dysfunction: orthostatic hypotension; incontinence; sexual dysfunction (Box 7.12)
Box 7.12
Fluctuating cognitive abilities and autonomic dysfunction together account for episodes of loss of consciousness.
c. visual hallucinations (80%); also illusions and paranoid delusions (Box 7.13)
i. hallucinations may be caused by intrusion of REM sleep while the patient is awake (i.e., as in peduncular hallucinosis)
d. sleep disturbance, particularly REM (rapid eye movements) sleep behavioral disorder (RSBD) that may precede the dementia by decades
i. conversely, an unspecified dementia occurring in conjunction with RSBD is likely to be Lewy body disease
e. impairment of visuospatial abilities without ophthalmologic abnormality (Box 7.14)
Box 7.14
Impairment of visuospatial abilities and parkinsonism together may account for frequent falls.
f. Parkinsonism: symptoms are symmetric and poorly responsive to dopaminergic agonists; tremor is mild and often involves a positional as well as resting component
i. the parkinsonism must develop no sooner than 1 year before the dementia to be diagnosed as Lewy body disease and not as Parkinson’s disease dementia
4. Diagnostic testing
a. polysomnography may demonstrate RSBD (i.e., the lack of atonia during REM sleep)
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


