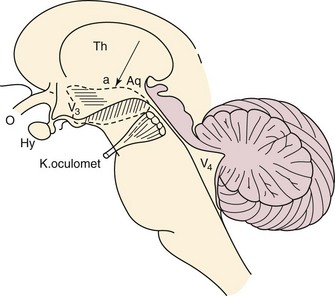
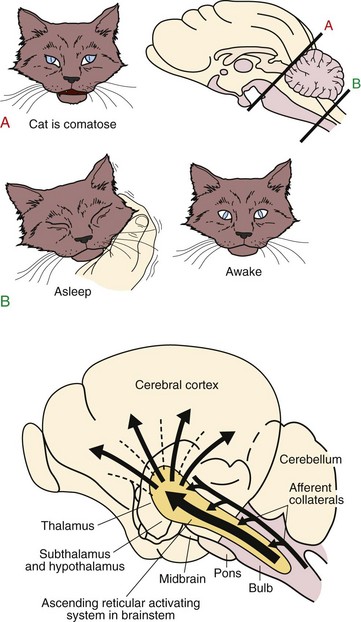
Chapter 3 3.1 Sleep Mechanisms Although it is generally believed that in sleep, the entire brain is “asleep,” research suggests that sleep may also be a localized process and that the whole brain is not “asleep” at the same time. What is clear is that sleep is an active process, whose timing and length are controlled by structures in the nervous system (Figs. 3.1-1 and 3.1-2). Figure 3.1-1 The basic circuitry, including key “nodes,” that underlies the regulation of sleep and wakefulness and the transitions between these states in mammals. Figure 3.1-2 An active role for the brain in sleep-wake behavior was first indicated in 1916, when Baron Constantine von Economo performed a postmortem brain analysis on victims of a viral encephalitis—encephalitis lethargica, or von Economo’s “sleeping sickness”—that profoundly affected sleep-wake regulation. As seen here in the original drawing taken from von Economo’s clinicoanatomic studies, lesions at the junction of the midbrain and posterior hypothalamus (diagonal hatching) produced hypersomnolence. By contrast, lesions of the basal forebrain and anterior hypothalamus (horizontal hatching) produced profound insomnia. Von Economo also observed that lesions between these two sites (arrow), which included the lateral hypothalamic area, caused narcolepsy. O, optic nerve (second cranial nerve); VE, third ventricle; Hy, hypothalamus; Th, thalamus; V4, fourth ventricle; Aq, cerebral aqueduct; K, oculomet, oculomotor nerve (third cranial nerve). (From Von Economo C: Sleep as a problem of localization. J Nerv Ment Dis 1930;71:249–259.) For many years, the nature of the circuitry that subserves the brain regions that promote wake/arousal and sleep remained elusive. The concept evolved that sleep control uses centers and processes that cause arousal and those that promote sleep (Figs. 3.1-3 and 3.1-4; see also Fig. 3.1-1). Figure 3.1-3 Top, In 1935, Bremer uncovered evidence of an ascending arousal system necessary for cortical arousal when he demonstrated that transection of the brainstem at the pontomesencephalic level (cerveau isole, A), but not the spinomedullary junction (encephale isole, B), produced coma in anesthetized cats. Bremer hypothesized that the resulting reduction in “cerebral tone” following the cerveau isole was due to interruption of ascending sensory inputs, that is, a passive deafferentation theory of sleep. Bottom, More than a decade after Bremer’s transection experiments, Moruzzi, a student of Bremer’s, and Magoun demonstrated that electrical stimulation of the rostral pontine reticular formation produced a desynchronized electroencephalogram, an electrophysiologic correlate of the conscious state, in anesthetized cats. Moruzzi and Magoun interpreted their experimental data as evidence for an active “waking center” in the mesopontine reticular formation, essentially refuting the deafferentation theory of sleep. Moruzzi and Magoun called this brainstem system the ascending reticular activating system. (Top, Modified from Bremer F: Bulletin de l’Académie Royale de Belgique 1937;4.) Figure 3.1-4 In the 1970s and 1980s, the neurochemistry of several brainstem “arousal” centers was elaborated. Neurons of the ventrolateral preoptic (VLPO) system and adjacent median preoptic nuclei (MnPO) are sleep-active neurons. Loss of VLPO neurons produces profound insomnia and sleep fragmentation (Figs. 3.1-5 and 3.1-6). Figure 3.1-5 The main sleep-promoting pathways from the ventrolateral preoptic nuclei (VLPO) and median preoptic nucleus (MnPO) inhibit the components of the ascending arousal pathways in both the hypothalamus and the brainstem, including awake-active noradrenergic neurons of the ventrolateral medulla and locus coeruleus (LC), cholinergic neurons in the pedunculopontine tegmental/laterodorsal tegmental nuclei, serotonergic neurons in the dorsal raphe nucleus, dopaminergic neurons of the ventral periaqueductal gray matter, histaminergic neurons of the tuberomammillary nucleus (TMN), and glutamatergic neurons of the parabrachial nucleus. This interaction between the VLPO and MnPO and components of the arousal system is, to a large degree, mutually inhibitory, and as such these pathways function analogously to an electronic “flip” switch/circuit. By virtue of the self-reinforcing nature of these switches—that is, when each side is firing, it reduces its own inhibitory feedback—the flip switch is inherently stable in either end state, but it avoids intermediate states. The flip design thus ensures stability of the behavioral state and facilitates rapid switching between behavioral states. The LH (lateral hypothalamic) orexin (hypocretin) neurons likely play a stabilizing role for the switch. Specifically, it appears that LH orexin neurons actively reinforce monoaminergic arousal tone. The position of the orexin neurons outside the flip switch permits them to stabilize the behavioral state by reducing transitions during both sleep and wakefulness. Narcoleptic humans or animals that lack orexin have increased transitions in both states. VLPOex, extended ventrolateral preoptic nucleus; VLPOc, core ventrolateral preoptic nucleus. (Modified from Fuller PM, Gooley JJ, Saper CB: Neurobiology of the sleep-wake cycle: sleep architecture, circadian regulation, and regulatory feedback. J Biol Rhythms 2006; 21[6]:482–493.) Figure 3.1-6 During cortical arousal, the electroencephalogram directly reflects the collective synaptic potentials of inputs, largely to pyramidal cells within the neocortex and hippocampus. Although significant progress has been made in delineating the neuronal circuitry that controls wakefulness and sleep, the basis of “sleep drive” has remained elusive. Sleep drive has been conceptualized as a homeostatic pressure that builds during the waking period and is dissipated by sleep. This homeostatic process, or “sleep homeostat,” thus represents the need for sleep (Fig. 3.1-7). Figure 3.1-7 The cellular determinant of homeostatic sleep drive is unknown, although a putative endogenous somnogen, adenosine, is believed to play a critical role. Rapid eye movement (REM) sleep is a distinct state in which the function of the central nervous system and the autonomic nervous system differs from both wakefulness and non-REM (NREM) sleep (Fig. 3.1-8). Figure 3.1-8 In this contemporary model of rapid eye movement (REM) sleep and non-REM sleep switching, two populations of mutually inhibitory neurons in the upper pons form a switch for controlling transitions between REM and NREM (A) sleep. In mammals, the circadian clock in the suprachiasmatic nuclei (SCN) of the anterior hypothalamus is critical for establishing the circadian rhythm of sleep-wake cycles. Regulation of sleep and wakefulness by the SCN is evident because the sleep-wake cycle continues on an approximately 24-hour basis, even when environmental conditions are constant (i.e., in the absence of environmental time cues), but only if the SCN is intact. In humans, a clear circadian variation in sleep propensity and sleep structure has been demonstrated by uncoupling the rest-activity cycle from the output of the circadian pacemaker (Fig. 3.1-9). Figure 3.1-9 The circadian rhythm of sleep-wake cycles is likely regulated at multiple levels in the hypothalamus. Anaclet, C, Lin, JS, Vetrivelan, R, et al. Identification and characterization of a sleep-active cell group in the rostral medullary brainstem. J Neurosci. 2012; 32(50):17970–17976. Basheer, R, Strecker, RE, Thakkar, MM, McCarley, RW. Adenosine and sleep-wake regulation. Prog Neurobiol. 2004; 73(6):379–396. Blanco-Centurion, C, Xu, M, Murillo-Rodriguez, E, et al. Adenosine and sleep homeostasis in the basal forebrain. J Neurosci. 2006; 26(31):8092–8100. Bremer, F. Cerveau “isole” et physiologie du sommeil. Comptes Rendus de la Société de Biologie (Paris). 1935; 118:1235–1241. Bremer, F. Preoptic hypnogenic area and reticular activating system. Arch Ital Biol. 1973; 111:85–111. Edgar, DM, Dement, WC, Fuller, CA. Effect on SCN lesions on sleep in squirrel monkeys: processes in sleep-wake regulation. J Neurosci. 1993; 13(3):1065–1079. Fuller, PM, Sherman, D, Pedersen, NP, Saper, CB, Lu, J. Reassessment of the structural basis of the ascending arousal system. J Comp Neurol. 2011; 519(5):933–956. Gerashchenko, D, Wisor, JP, Burns, D, et al. Identification of a population of sleep-active cerebral cortex neurons. Proc Natl Acad Sci U S A. 2008; 105(29):10227–10232. Lu, J, Sherman, D, Devor, M, Saper, CB. A putative flip-flop switch for control of REM sleep. Nature. 2006; 441(7093):589–594. Morairty, S, Rainnie, D, McCarley, R, Greene, R. Disinhibition of ventrolateral preoptic area sleep-active neurons by adenosine: a new mechanism for sleep promotion. Neuroscience. 2004; 123(2):451–457. Moruzzi, G, Magoun, H. Brainstem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol. 1949; 1:455–473. Nauta, WJH. Hypothalamic regulation of sleep in rats: experimental study. J Neurophysiol. 1946; 9:285–316. Sherin, JE, Elmquist, JK, Torrealba, F, Saper, CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998; 18(12):4705–4721. Sherin, JE, Shiromani, PJ, McCarley, RW, Saper, CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996; 271(5246):216–219. Steriade, M, McCormick, DA, Sejnowski, TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993; 262:679–685. Von Economo, C. Sleep as a problem of localization. J Nerv Ment Dis. 1930; 71:249–259. 3.2 Localization and Neurochemistry of Sleep-Wake Physiology and Pathophysiology Figure 3.2-1 shows the network responsible for the waking state. Waking state (Wk-on) neurons activate the cortex either directly or by means of a relay in the intralaminar thalamic nuclei.1–3 Direct pathways from the noradrenergic and serotonergic neurons to the cortex contributing to the waking state are not represented for sake of clarity of the figure. During the waking state, REM-off GABAergic neurons located in the vlPAG and the dorsal part of the deep mesencephalic nucleus (dDPMe) tonically inhibit SLD glutamatergic neurons. These REM-off GABAergic neurons are excited by monoaminergic and hypocretin inputs. Figure 3.2-1 Network responsible for the waking state. In contrast to the complex and extensive neurochemical network involved in waking, the neurons inducing NREM sleep are localized in the lateral preoptic area and the adjacent basal forebrain (BF; Fig. 3.2-2). As previously mentioned, a cluster of these neurons is localized in the VLPO. It has been demonstrated in rats that lesions including the VLPO induce insomnia for up to several weeks.4 The GABAergic neurons of the VLPO are active specifically during slow-wave sleep.5,6 Sherin and colleagues7 identified a population of neurons in the VLPO that showed expression of the proto-oncogene c-Fos, a marker of neuronal activation following recovery sleep in previously sleep-deprived rats.8 Figure 3.2-2 Network responsible for non–rapid eye movement (REM) sleep. Based on all data available, a reciprocal interaction of wake-promoting and NREM sleep-promoting neurons across the sleep-wake cycle has been suggested. This theory is supported by in vitro pharmacologic studies in rat brain slices that revealed that VLPO neurons are indeed inhibited by most of the waking transmitters.9 VLPO and MnPo neurons would be tonically inhibited by noradrenergic and cholinergic inputs during waking. At the onset of sleep, the GABAergic sleep neurons of the VLPO and the MnPo would be activated by the circadian clock, which is localized in the SCN, and the hypnogenic factor adenosine, which progressively accumulates in the brain during waking. The effect of this accumulation of adenosine is the target for one of the most commonly used psychoactive drugs, caffeine,10 which acts via antagonism of the A1 and A2A adenosine receptors. In turn, these sleep-active neurons begin to inhibit the wake-active neurons in the multiple arousal centres via the neurotransmitter GABA, leading to synchronization of the thalamocortical network. The decrease in sensory inputs is also necessary to let sleep occur since waking systems are excited by them. It was first shown that REM sleep persists after decortication, cerebellar ablation, or brainstem transections rostral to the pons. In contrast, transection at the posterior limit of the pons suppressed REM sleep.11 It was then demonstrated that a state resembling REM sleep is still visible in the “pontine cat,” a preparation in which all the structures rostral to the pons have been removed (Fig. 3.2-3).11 These results indicated that brainstem structures are necessary and sufficient to trigger and maintain the state of REM sleep, a concept still valid today. By using electrolytic and chemical lesions, it was then evidenced that the dorsal part of the pontis oralis (PnO) and caudalis (PnC) nuclei contains the neurons responsible for REM sleep onset.11 Furthermore, bilateral injections in cats of a cholinergic agonist (carbachol) into the dorsal area of PnO and PnC (also named peri-locus coeruleus-α), pontine inhibitory area, subcoeruleus nucleus, and, more recently, the SLD in rats dramatically increases REM sleep quantities in cats.12 It was then shown by unit recordings in freely moving cats that many SLD neurons show a tonic firing selective to REM sleep (called “REM-on” neurons).12 It was believed that SLD REM-on neurons are cholinergic until we recently showed that they are glutamatergic.12 Figure 3.2-3 Localization of the pontine generator responsible for rapid eye movement sleep (paradoxical [PS] sleep). REM sleep onset is due to the activation of glutamatergic REM-on neurons localized in the SLD (Fig. 3.2-4). During wakefulness and NREM sleep, these REM-on neurons are inhibited by a tonic inhibitory GABAergic tone originating from REM-off neurons localized in the vlPAG and the dDpMe. These neurons would be activated during wakefulness by the hypocretin (orexin)-containing (Hcrt) neurons and the monoaminergic neurons. The onset of REM sleep is due to the activation by intrinsic mechanisms of REM-on GABAergic neurons localized in the posterior lateral hypothalamic area, the dorsal paragigantocellular reticular nucleus (DPGi), and the vlPAG. These neurons also inactivate the REM-off monoaminergic neurons during REM sleep. The disinhibited ascending SLD REM-on neurons in turn induce cortical activation via their projections to intralaminar thalamic relay neurons in collaboration with wakefulness/REM-on cholinergic and glutamatergic neurons from the laterodorsal tegmental nucleus and pedunculopontine nucleus, mesencephalic and pontine reticular nuclei, and the BF. Descending REM-on SLD neurons induce muscle atonia via their excitatory projections to glycinergic premotoneurons localized in the alpha and ventral gigantocellular reticular nuclei (Gia and GiV, respectively). The exit from REM sleep is due to the activation of waking systems because REM sleep episodes are almost always terminated by an arousal. The waking systems inhibit all GABAergic REM-on neurons. Because the duration of REM sleep is negatively coupled with the metabolic rate, we propose that the activity of the waking systems is triggered to end REM sleep to restore competing physiologic parameters, such as thermoregulation.12 Figure 3.2-4 Network responsible for rapid eye movement sleep (REM). In idiopathic REM behavior disorder patients, the descending, but not the ascending, SLD glutamatergic neurons may have degenerated (Fig. 3.2-5). Another possibility is that only the GiV glycinergic/GABAergic neurons did degenerate. Movements are induced during REM sleep by direct or indirect glutamatergic projections from the motor cortex to spinal and cranial motor neurons.12 Figure 3.2-5 Network responsible for rapid eye movement (REM) sleep during idiopathic REM sleep behavior disorder (RBD). In narcoleptics, the network responsible for muscle atonia is intact (Fig. 3.2-6). Phasic movements during REM sleep are induced by a phasic activation of motoneurons due to the absence of an excitatory projection of the orexin-containing neurons to the descending SLD REM-on neurons.12 Figure 3.2-6 Network responsible for rapid eye movement (REM) sleep during REM sleep disorder (RBD) in narcoleptics. Cataplexy is induced by the activation of a direct or indirect pathway from the emotionally driven central amygdala to the descending SLD glutamatergic neurons (Fig. 3.2-7). In healthy subjects, the hypocretin neurons are excited. They excite GABAergic PS-off neurons located in the vlPAG/dDPMe and, by this means, increase the inhibition of SLD glutamatergic neurons and counteract the excitation coming from the central amygdala.12 Figure 3.2-7 Network responsible for rapid eye movement (REM) sleep during cataplexy in narcoleptics. 1. Moruzzi, G, Magoun, HW. Brain stem reticular formation and activation of the EEG. J Neuropsychiatry Clin Neurosci. 1949; 7(2):251–267. 2. Fort, P, Bassetti, CL, Luppi, PH. Alternating vigilance states: new insights regarding neuronal networks and mechanisms. Eur J Neurosci. 2009; 29(9):1741–1753. 3. Jones, BE. Basic mechanisms of sleep-wake states. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medecine. Philadelphia: Saunders, W. B. ; 1994:145–162. 4. Lu, J, Greco, MA, Shiromani, P, Saper, CB. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci. 2000; 20(10):3830–3842. 5. Sherin, JE, Elmquist, JK, Torrealba, F, Saper, CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998; 18(12):4705–4721. 6. Szymusiak, R, Alam, N, Steininger, TL, McGinty, D. Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res. 1998; 803(1–2):178–188. 7. Sherin, JE, Shiromani, PJ, McCarley, RW, Saper, CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996; 271(5246):216–219. 8. Morgan, JI, Curran, T. Stimulus-transcription coupling in the nervous system: involvement of the inducible proto-oncogenes fos and jun. Annu Rev Neurosci. 1991; 14:421–451. 9. Gallopin, T, Fort, P, Eggermann, E, Cauli, B, Luppi, PH, Rossier, J, et al. Identification of sleep-promoting neurons in vitro. Nature. 2000; 404(6781):992–995. 10. Fredholm, BB, Chen, JF, Masino, SA, Vaugeois, JM. Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol. 2005; 45:385–412. 11. Jouvet, M. Recherches sur les structures nerveuses et les mécanismes responsables des différentes phases du sommeil physiologique. Arch Ital Biol. 1962; 100:125–206. 12. Luppi, PH, Clement, O, Sapin, E, Gervasoni, D, Peyron, C, Leger, L, et al. The neuronal network responsible for paradoxical sleep and its dysfunctions causing narcolepsy and rapid eye movement (REM) behavior disorder. Sleep Med Rev. 2011; 15(3):153–163. 3.3 Circadian Rhythms Regulation All living creatures exhibit self-sustaining circadian (~24 hour) rhythms in physiology and behavior that are regulated by a central “clock” located in the suprachiasmatic nuclei (SCN) of the hypothalamus. This system is involved in the regulation of many physiologic functions that include core body temperature (CBT), sleep, and the timing of the production of hormones. The genetics of the circadian system at the cellular level has only recently been understood (Fig. 3.3-1). The central circadian clock, the SCN, also controls the timing of peripheral clocks in the liver, heart, and adrenal glands, for example, and these peripheral rhythms may then in turn feed back to the SCN (Fig. 3.3-2). Figure 3.3-1 The genetic mechanism of the circadian clock is similar in many species, from prokaryotic bacteria to fruit flies to mammals. Figure 3.3-2 Communication routes of the mammalian circadian timing system. The sleep-wake cycle is the most apparent circadian rhythm in humans. Light, physical activity, and melatonin from the pineal gland are the primary synchronizing agents for the human circadian clock. In humans, light is the strongest time cue for the circadian clock. For optimal function, the timing of socially or work-imposed sleep and wake schedules should be designed and taken into account with the external physical environment and the circadian rhythms. Circadian rhythm sleep disorders (see Chapter 9) develop when the circadian clock or its entraining pathways are disrupted, or when the external environment is misaligned with the timing of the circadian clock (Figs. 3.3-3 and 3.3-4). Figure 3.3-3 The basic components of the circadian system. Figure 3.3-4 At least two variables seem to play a role in the regulation of the timing of sleep.
Biology of Sleep
Nervous System Control
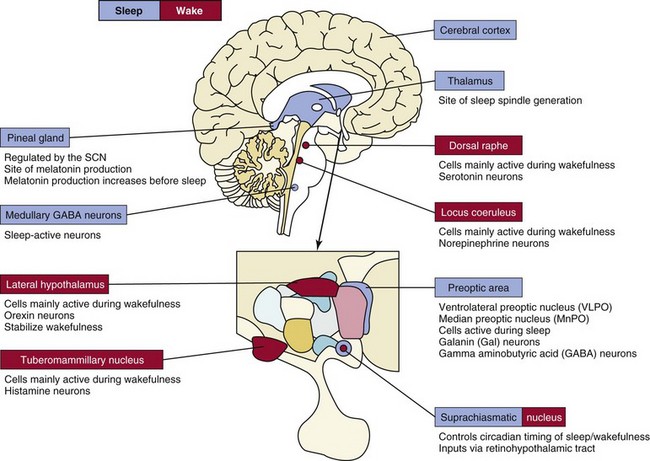
Current models of wakefulness and sleep hold that wakefulness is maintained by the combined excitatory influence of forebrain-projecting noradrenergic (locus coeruleus), histaminergic (tuberomammillary nucleus), serotoninergic (dorsal raphe), and cholinergic (not shown) cell groups located at or near the mesopontine junction. Sleep, on the other hand, is initiated and maintained by neurons in the median preoptic (MnPO) and ventrolateral preoptic (VLPO) nuclei via inhibitory projections to the more rostrally situated wakefulness-promoting cell groups. Hypocretin (orexin) neurons located in the lateral hypothalamus reinforce activity in the brainstem arousal pathways and also stabilize both sleep and wakefulness. Disruption of the hypocretin system leads to narcolepsy. The suprachiasmatic nuclei (SCN) determine the timing of the sleep-wake cycle and help to “consolidate” these behavioral states. The pineal gland, located in the epithalamus, produces melatonin, a hormone thought to function as a hypnotic signal. The cerebral cortex and medullary brainstem also contain subpopulations of γ-amino butyric acid (GABA)-ergic sleep-active neurons.
Arousal Systems
A, In the contemporary view, the ascending arousal system consists of noradrenergic (NE) neurons of the ventrolateral medulla and locus coeruleus (LC), cholinergic neurons (ACh) in the pedunculopontine tegmental/laterodorsal tegmental (PPT/LDT) nuclei, serotonergic (5-hydroxytryptamine [5-HT]) neurons in the dorsal raphe nucleus, dopaminergic neurons (DA) of the ventral periaqueductal gray matter (vPAG), and histaminergic neurons (His) of the tuberomammillary nucleus (TMN). Wakefulness-promoting glutamatergic neurons of the parabrachial nucleus also provide direct innervation of the hypothalamus, basal forebrain (BF), and cerebral cortex. These systems collectively produce cortical arousal via two pathways: a dorsal route through the thalamus and a ventral route through the hypothalamus and BF. The latter pathway receives contributions from the hypocretin (orexin, ORX) and melanin-concentrating hormone (MCH) neurons of the lateral hypothalamic area (LHA) as well as from γ-amino butyric acid (GABA)-ergic or cholinergic neurons of the BF. Note that all these ascending pathways traverse the region at the midbrain-diencephalic junction, where von Economo observed that lesions caused hypersomnolence. As shown here, several putative brainstem arousal centers were identified and characterized nearly 30 years ago. It nevertheless remained unclear for many years how this arousal system was “turned off” so that sleep could be initiated and maintained. B, Although work by W.J.A. Nauta in 1946 provided support for the concept of sleep-promoting circuitry in the anterior hypothalamus/preoptic area, it was not until the mid-1990s that the identity of this sleep-promoting circuitry was revealed. In these investigations, it was demonstrated that the median preoptic nucleus and ventrolateral preoptic nucleus (VLPO) contain sleep-active cells, the latter of which contains the inhibitory neurotransmitters GABA and galanin (Gal). The VLPO (open orange circle) projects to all the main components of the ascending arousal system. Inhibition of the arousal system by the VLPO during sleep is critical for the maintenance and consolidation of sleep. Recent work has revealed a node of GABAergic neurons in the parafacial zone of the rostral medulla that are also sleep active.
Sleep-Promoting Systems
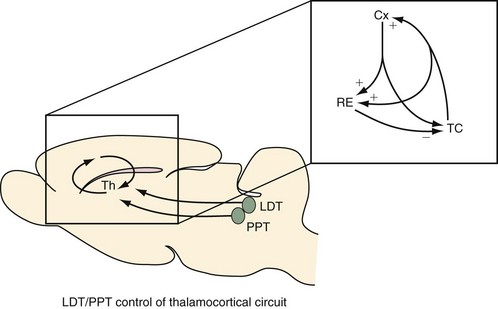
The thalamocortical (TC) system has been widely considered to be a major source of this activity. In turn, the overall level of activity in the TC system is thought to be regulated by the ascending arousal system. Today, it is generally accepted that a brainstem cholinergic activating system, located in the pedunculopontine tegmental (PPT) and laterodorsal tegmental (LDT) nuclei (green circles), induces tonic and phasic depolarization effects on TC neurons to produce the low-voltage, mixed-frequency, fast activity of the waking and rapid eye movement (REM) sleep electroencephalogram (EEG). The PPT and LDT cease firing during non-REM (NREM) sleep, which hyperpolarizes the TC neurons to produce two important effects: 1) sensory transmission through the thalamus (Th) to the cortex is blocked; and 2) oscillatory activity between TC neurons, cortical neurons (Cx), and reticular thalamus (RE) neurons (inset) is unmasked to manifest several characteristics of the sleep EEG—slow-wave activity (0.5 to 4 Hz) and spindles. Thus the thalamus appears to be a critical relay for the ascending arousal system for “gating” sensory transmission to the cortex during sleep and wakefulness. In addition to the thalamus, a critical “relay” role for the basal forebrain in maintaining electrocortical and neurobehavioral arousal has also recently been demonstrated.
Sleep Drive
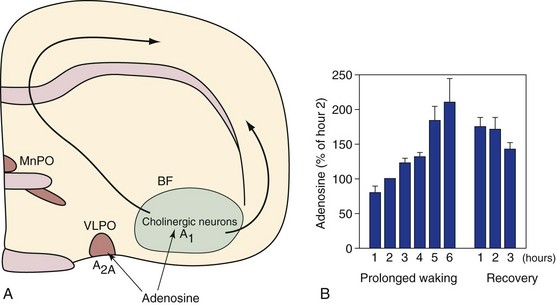
A, Adenosine is a naturally occurring purine nucleoside. It is hypothesized that adenosine accumulates during wakefulness, and upon reaching sufficient concentrations, it inhibits neural activity in wake-promoting circuitry of the basal forebrain (BF) via A1 receptors located on BF cholinergic neurons. It is likely that it activates sleep-promoting ventrolateral preoptic nuclei (VLPO) neurons, via A2A receptors, located adjacent to the BF. A role for adenosine and BF cholinergic neurons in sleep homeostasis has recently been challenged by Blanco-Centurion and colleagues. B, Mean BF extracellular adenosine values by hour during 6 hours of prolonged wakefulness and in the subsequent 3 hours of spontaneous recovery sleep in cats. Microdialysis values in the six animals are normalized relative to the second hour of wakefulness. MnPO, median preoptic nucleus. (Data from Porkka-Heiskanen T, Strecker RE, Thakkar M, et al: Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science 1997;276[5316]:1265–1268.)
Control of Rapid Eye Movement Sleep
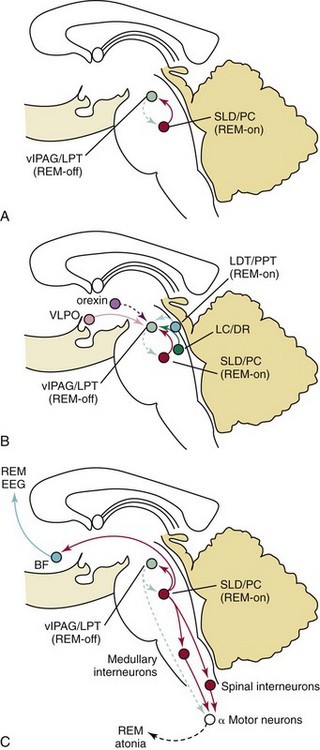
γ-Aminobutyric acid (GABA)-ergic ventrolateral periaqueductal gray matter (vlPAG, light green) neurons and laterodorsal tegmental/pedunculopontine (LPT/PPT, light green) neurons fire during non-REM states to inhibit entry into REM sleep, during which these neurons are inhibited by a population of GABAergic REM sleep–active neurons in the sublaterodorsal region (SLD; red). This mutually inhibitory relationship produces a REM-NREM flip switch, promoting rapid and complete transitions between these states. The core REM sleep switch is, in turn, modulated by other neurotransmitter systems (B). Noradrenergic neurons in the locus coeruleus (LC) and serotonergic neurons in the dorsal raphe (DR; dark green) inhibit REM sleep by actions on both sides of the flip switch (exciting REM-off neurons and inhibiting REM-on neurons); during REM sleep, they are silent (dashed lines), whereas cholinergic neurons (blue) promote REM sleep by having opposite actions on the same two neuronal populations. The hypercretin (orexin) neurons inhibit entry into REM sleep by exciting neurons in the REM-off population (and by presynaptic effects that excite monoaminergic terminals), whereas the ventrolateral preoptic nuclei (VLPO) neurons promote entry into REM sleep by inhibiting this same target. During REM sleep (C), a separate population of glutamatergic neurons in the SLD (red) activates a series of inhibitory interneurons in the medulla and spinal cord, which inhibit motor neurons and thereby produce the atonia of REM sleep. Withdrawal of tonic excitatory input from the REM-off regions may also contribute to the loss of muscle tone. At the same time, ascending projections from glutamatergic neurons in the parabrachial (PB) nucleus and pre-coeruleus (PC) area activate forebrain pathways that drive EEG desynchronization and hippocampal theta rhythms, thus producing the characteristic EEG signs of REM sleep. BF, basal forebrain; EEG, electroencephalogram. (Modified from Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE: Sleep state switching. Neuron 2010;68[6]:1023–1042.)
Control of the Timing of Sleep
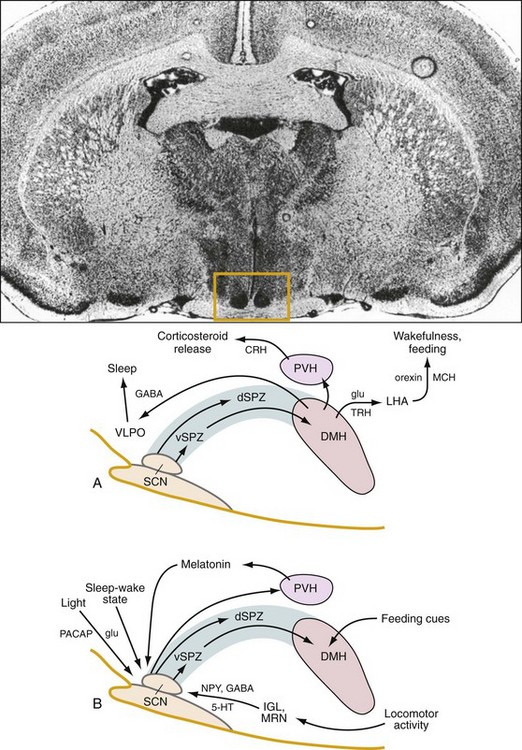
Top, A coronal section of the brain shows the location (box) of the rat suprachiasmatic nucleus (SCN). A, The circadian clock in the SCN sends an indirect projection to the dorsomedial hypothalamic nucleus (DMH) via the subparaventricular zone (SPZ), which is critical for the circadian rhythm of sleep-wake cycles. The DMH, in turn, provides rhythmic output to brain regions critical for the regulation of sleep and wakefulness, hormone synthesis and release, and feeding. B, This multistage regulation of circadian behavior in the hypothalamus allows for the integration of multiple time cues from the environment to shape daily patterns of sleep and wakefulness. 5-HT, 5-hydroxytryptamine (serotonin); CRH, corticotrophin-releasing hormone; dSPZ, dorsal subparaventricular zone; GABA, γ-amino butyric acid; glu, glutamate; IGL, intergeniculate leaflet; LHA, lateral hypothalamic area; MCH, melanin-concentrating hormone; MRN, median raphe nucleus; NPY, neuropeptide Y; PACAP, pituitary adenylate cyclase-activating polypeptide; PVH, paraventricular hypothalamic nucleus; TRH, thyrotropin-releasing hormone; VLPO, ventrolateral preoptic nucleus; vSPZ, ventral subparaventricular zone. (Modified from Fuller PM, Gooley JJ, Saper CB: Neurobiology of the sleep-wake cycle: sleep architecture, circadian regulation, and regulatory feedback. J Biol Rhythms 2006;21[6]:482–493.)
Explanatory Diagrams
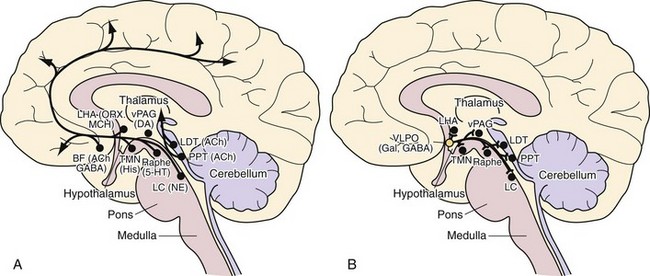
The Waking State
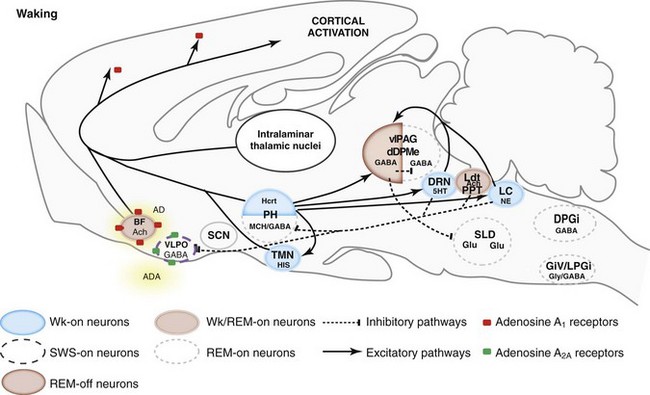
5HT, serotonin; AD, adenosine; Ach, acetylcholine; BF, basal forebrain; DPGi, dorsal paragigantocellular reticular nucleus; dDpMe, dorsal deep mesencephalic reticular nucleus; DRN, dorsal raphe nucleus; GABA, γ-aminobutyric acid; GiV, ventral gigantocellular reticular nucleus; Glu, glutamate; Gly, glycine; HIS, histamine; Hcrt, hypocretin (orexin)-containing neurons; LC, locus coeruleus; Ldt, laterodorsal tegmental nucleus, LPGi, lateral paragigantocellular reticular nucleus; MCH, melanin-concentrating hormone; NE, norepinephrine; PH, posterior hypothalamus; PPT, pedunculopontine nucleus; REM, rapid eye movement sleep; SCN, suprachiasmatic nucleus; SLD, sublaterodorsal nucleus; SWS, slow-wave sleep; TMN, tuberomammillary nucleus; vlPAG, ventrolateral periaqueductal gray matter; VLPO, ventrolateral preoptic nucleus; Wk, wakefulness.
NREM Sleep
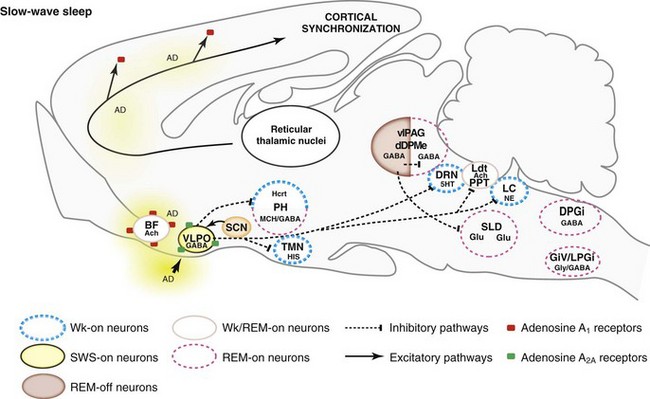
5HT, serotonin; AD, adenosine; Ach, acetylcholine; BF, basal forebrain; DPGi, dorsal paragigantocellular reticular nucleus; dDpMe, dorsal deep mesencephalic reticular nucleus; DRN, dorsal raphe nucleus; GABA, γ-aminobutyric acid; GiV, ventral gigantocellular reticular nucleus; Glu, glutamate; Gly, glycine; Hcrt, hypocretin- (orexin) containing neurons; HIS, histamine; LC, locus coeruleus; Ldt, laterodorsal tegmental nucleus, LPGi, lateral paragigantocellular reticular nucleus; MCH, melanin-concentrating hormone; NE, norepinephrine; PH, posterior hypothalamus; PPT, pedunculopontine nucleus; SCN, suprachiasmatic nucleus; SLD, sublaterodorsal nucleus; SWS, slow-wave sleep; TMN, tuberomammillary nucleus; vlPAG, ventrolateral periaqueductal gray matter; VLPO, ventrolateral preoptic nucleus; Wk, wakefulness.
Pontine Generator in REM Sleep
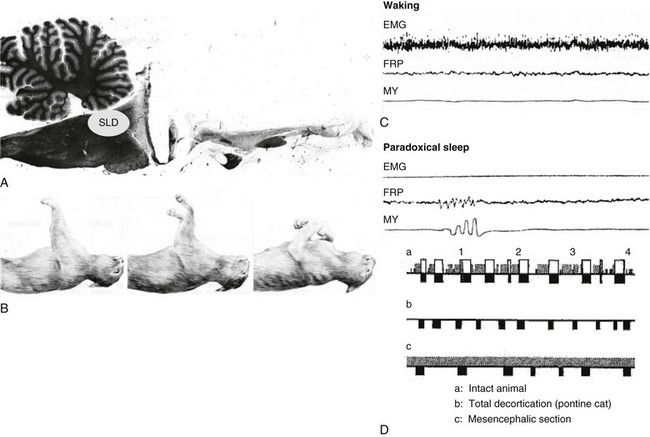
A, Sagittal section of the brain of a pontine cat. B, During PS, the cats display atonia in contrast during waking, as shown in the two photos on the left. C, Recordings of the electromyogram (EMG), local field potential in the pontine reticular formation (FRP), and ocular movements (MY) during waking and paradoxical sleep. D, Hypnograms of normal (a), pontine (b), and sectioned (c) cats. SLD, sublaterodorsal tegmental nucleus. (Modified from Jouvet M: Research on the neural structures and responsible mechanisms in different phases of physiological sleep [in Italian]. Arch Ital Biol 1962;100:125–206.)
Network Responsible for REM Sleep
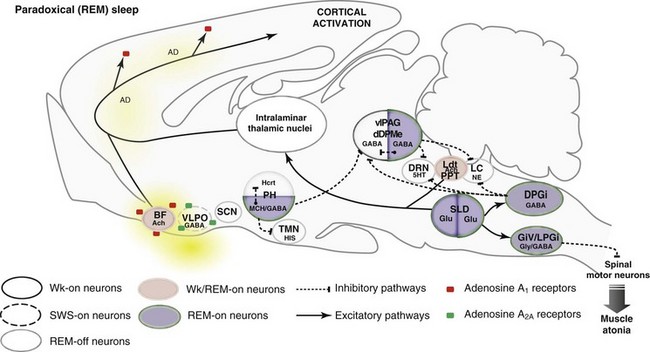
5HT, serotonin; AD, adenosine; Ach, acetylcholine; BF, basal forebrain; DPGi, dorsal paragigantocellular reticular nucleus; dDpMe, dorsal deep mesencephalic reticular nucleus; DRN, dorsal raphe nucleus; GABA, γ-aminobutyric acid; GiV, ventral gigantocellular reticular nucleus; Glu, glutamate; Gly, glycine; Hcrt, hypocretin- (orexin) containing neurons; HIS, histamine; LC, locus coeruleus; Ldt, laterodorsal tegmental nucleus, LPGi, lateral paragigantocellular reticular nucleus; MCH, melanin concentrating hormone; NE, norepinephrine; PH, posterior hypothalamus; PPT, pedunculopontine nucleus; SCN, suprachiasmatic nucleus; SLD, sublaterodorsal nucleus; SWS, slow-wave sleep; TMN, tuberomammillary nucleus; vlPAG, ventrolateral periaqueductal gray matter; VLPO, ventrolateral preoptic nucleus; Wk, wakefulness.
Network Responsible for REM Sleep During Idiopathic REM Behavior Disorder
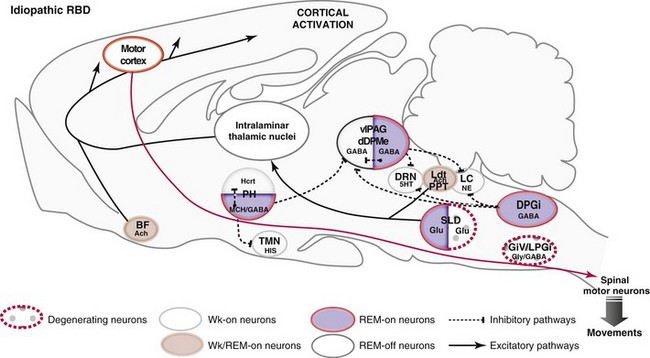
5HT, serotonin; Ach, acetylcholine; BF, basal forebrain; DPGi, dorsal paragigantocellular reticular nucleus; dDpMe, dorsal deep mesencephalic reticular nucleus; DRN, dorsal raphe nucleus; GABA, γ-aminobutyric acid; GiV, ventral gigantocellular reticular nucleus; Glu, glutamate; Gly, glycine; Hcrt, hypocretin- (orexin) containing neurons; HIS, histamine; LC, locus coeruleus; Ldt, laterodorsal tegmental nucleus, LPGi, lateral paragigantocellular reticular nucleus; MCH, melanin-concentrating hormone; NE, norepinephrine; PH, posterior hypothalamus; PPT, pedunculopontine nucleus; SLD, sublaterodorsal nucleus; TMN, tuberomammillary nucleus; vlPAG, ventrolateral periaqueductal gray matter; Wk, wakefulness.
Network Responsible for Paradoxical Sleep During REM Behavior Disorder in Narcoleptics
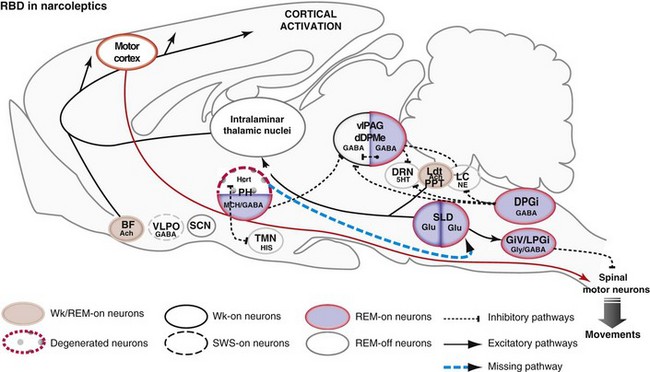
5HT, serotonin; Ach, acetylcholine; BF, basal forebrain; DPGi, dorsal paragigantocellular reticular nucleus; dDpMe, dorsal deep mesencephalic reticular nucleus; DRN, dorsal raphe nucleus; GABA, γ-aminobutyric acid; GiV, ventral gigantocellular reticular nucleus; Glu, glutamate; Gly, glycine; Hcrt, hypocretin (orexin)-containing neurons; HIS, histamine; LC, locus coeruleus; Ldt, laterodorsal tegmental nucleus, LPGi, lateral paragigantocellular reticular nucleus; MCH, melanin concentrating hormone; NE, norepinephrine; PH, posterior hypothalamus; PPT, pedunculopontine nucleus; SCN, suprachiasmatic nucleus; SLD, sublaterodorsal nucleus; SWS, slow-wave sleep; TMN, tuberomammillary nucleus; vlPAG, ventrolateral periaqueductal gray matter; VLPO, ventrolateral preoptic nucleus; Wk, wakefulness.
Network Responsible for Paradoxical Sleep During Cataplexy in Narcoleptics
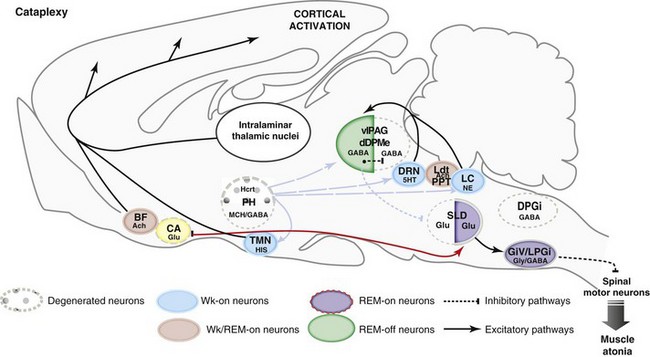
5HT, serotonin; Ach, acetylcholine; BF, basal forebrain; CA, central amygdala; DPGi, dorsal paragigantocellular reticular nucleus; dDpMe, dorsal deep mesencephalic reticular nucleus; DRN, dorsal raphe nucleus; GABA, γ-aminobutyric acid; GiV, ventral gigantocellular reticular nucleus; Glu, glutamate; Gly, glycine; Hcrt, hypocretin (orexin)-containing neurons; HIS, histamine; LC, locus coeruleus; Ldt, laterodorsal tegmental nucleus, LPGi, lateral paragigantocellular reticular nucleus; MCH, melanin concentrating hormone; NE, norepinephrine; PH, posterior hypothalamus; PPT, pedunculopontine nucleus; SLD, sublaterodorsal nucleus; TMN, tuberomammillary nucleus; vlPAG, ventrolateral periaqueductal gray matter; Wk, wakefulness.
References
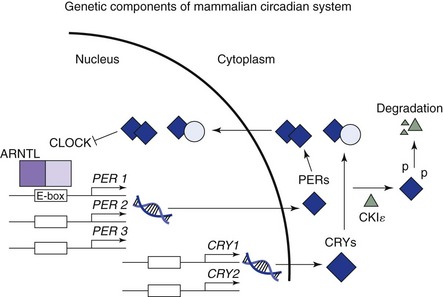
The key mechanism appears to involve an exquisitely timed transcription and translation feedback loop between nuclear gene expression and their protein products. In humans, dimers of PER and CRY proteins bind to dimers of CLOCK and ARNTL on the enhancer (E) box for PER genes and inhibit transcription of the PER and CRY genes. Reduced PER/CRY transcription to mRNAs and translation to proteins subsequently disinhibit CLOCK/ARNTL binding to the E-box, which leads to renewed transcription of PER and CRY.
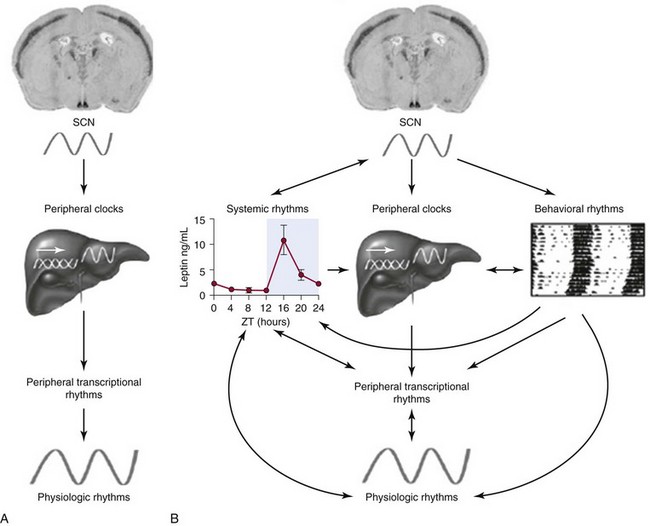
A, The suprachiasmatic nucleus (SCN) pacemaker entrains peripheral clocks. Via transcriptional regulation of clock-controlled genes in target tissues, peripheral physiologic functions are reset and synchronized to the light-dark cycle. B, Pathways of interaction between central and peripheral clocks. The SCN resets physiologic rhythms via the entrainment of peripheral tissue clocks, but at the same time regulates behavioral and systemic (e.g., endocrine) functions in a more direct manner. Peripheral rhythms may, in turn, directly or indirectly feed back to the SCN. This interlocked balance system creates plasticity in the entrainment of the circadian timing system and promotes adaptation to complex changes in environmental parameters. (From Barclay J, Tsang A, Oster H: Interaction of central and peripheral clocks in physiological regulation. Prog Brain Res 2012; 199:163–181.)
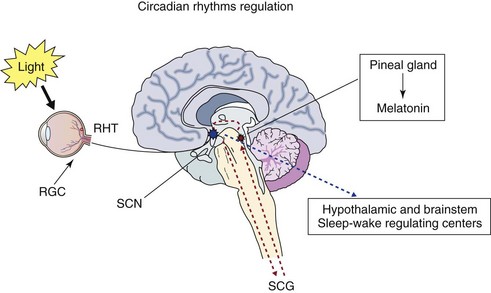
Photic information that reaches the suprachiasmatic nucleus (SCN) is transmitted from the retina via the retinohypothalamic tract (RHT). The retinal ganglion cells (RGC) of the eye, melanopsin-containing photoreceptors, provide the primary photic input to the circadian clock, transmitting the signal to the neurons of the SCN. Melatonin is released from the pineal gland at night, and its output is regulated by the SCN via the superior cervical ganglion (SCG). In addition to its ability to synchronize circadian rhythms, melatonin can also promote sleep. Integrated timing information from the SCN is transmitted to sleep-wake centers in the brain. Thus the sleep-wake cycle is generated by a complex interaction of endogenous circadian and sleep processes as well as by social and environmental factors.
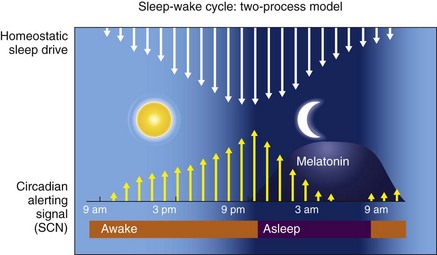
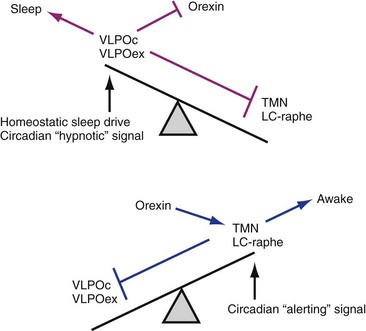
First is the homeostatic sleep drive, which increases the longer a person is awake. The second is timing information from the suprachiasmatic nucleus (SCN). In this two-process model, the SCN promotes wakefulness by stimulating arousal networks. SCN activity appears to oppose the homeostatic sleep drive, thus the alerting mediated by the SCN increases during the day. The propensity to be awake or asleep at any time is related to the homeostatic sleep drive and the opposing SCN alerting signal. At normal bedtime, both the alerting drive and the sleep drive are at their highest level. The SCN has at least two types of melatonin receptors, MT1 and MT2, involved in the regulation of sleep. Stimulation of MT1 receptors is believed to decrease the alerting signal from the SCN, whereas MT2 stimulation is thought to be involved in synchronizing the circadian system. (Data from Dijk DJ, Edgar DM: Circadian and homeostatic control of wakefulness and sleep. In Turek FW, Zee PC, editors: Regulation of sleep and circadian rhythms. New York: Marcel Dekker, 1999;111–147.)![]()
Stay updated, free articles. Join our Telegram channel







Full access? Get Clinical Tree


