and Cheng-Ying Ho2
(1)
Pathology, Johns Hopkins University, Baltimore, Maryland, USA
(2)
Pathology, Children’s National Medical Center, Washington, D.C., District of Columbia, USA
Diffuse Gliomas in Adults
Clinical and Pathologic Features
Diffuse gliomas represent the main category of intraparenchymal primary brain tumors in adults. According to the CBTRUS statistical report of primary brain tumors in the United States, gliomas correspond to 28 % of all brain and CNS tumors and 80 % of malignant ones, which are predominantly diffuse [1]. Histologic subtypes include astrocytomas and oligodendrogliomas and are graded in a scale from grade II–IV under the current WHO Classification.
Diffuse astrocytoma (WHO grade II) is considered a low-grade neoplasm, characterized mainly by cytologic atypia, with mitoses very rare to absent and lacking necrosis and microvascular proliferation. The cellularity of these tumors may be very low, and therefore, they may be difficult to distinguish from reactive conditions, e.g., gliosis, without the use of diagnostic biomarkers. Anaplastic astrocytomas (WHO grade III) are characterized by increased cellularity and mitotic activity, while glioblastoma (WHO grade IV), in addition to these features, also has microvascular proliferation and/or necrosis. Historically, a distinction has been made between primary (“de novo”) glioblastoma, the most common subtype, arising without an identifiable precursor, and secondary glioblastoma, developing from a preexisting lower-grade precursor. Although both subtypes are histologically indistinguishable, they have different molecular alterations (Figs. 5.1 and 5.2).
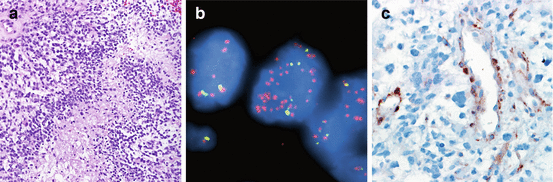
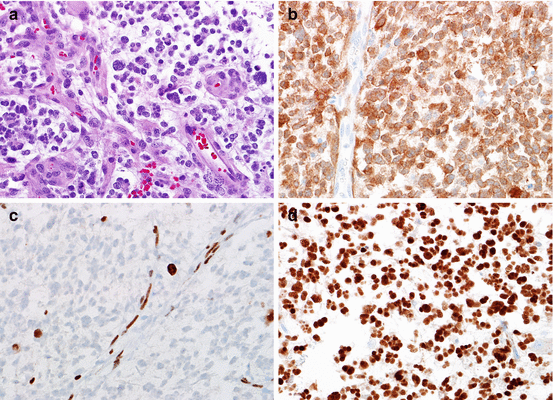
Fig. 5.1
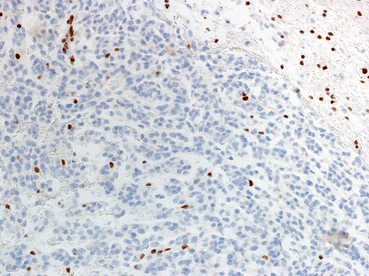
Genetic alterations in primary glioblastoma. At the histologic level, glioblastoma is defined by an infiltrating astrocytoma with mitotic activity and necrosis, frequently of the pseudopalisading type (a). Primary glioblastomas frequently have EGFR amplification, which is usually high level as demonstrated by FISH (EGFR red, CEP7 probe control green) (b). Alterations in chromosome 10 and loss of PTEN expression are also frequent. The latter may be detected by immunohistochemistry, where loss of expression is a feature of tumor cells, while nonneoplastic components (e.g., endothelium) show retained expression (c) (Color figure online)
Fig. 5.2
Biomarkers in secondary glioblastoma. Secondary glioblastomas are defined similar to primary glioblastomas, as infiltrating astrocytomas with mitotic activity and necrosis and/or microvascular proliferation as illustrated (a). However, at the molecular level, they are distinct and share similar alterations as lower-grade astrocytic neoplasms, including IDH1 (R132H) expression (b), ATRX loss in neoplastic cells (c), and strong p53 staining suggestive of TP53 mutations (d). This molecular signature is associated with a more favorable prognosis in diffuse astrocytic neoplasms
Oligodendroglioma (WHO grade II), representing the lower grade end of the spectrum, usually has low to moderate cellularity and low mitotic activity. These tumors contain round, uniform nuclei, and frequently perinuclear halos (Fig. 5.3). However, a greater level of mitotic activity may be tolerated in these tumors before contemplating a diagnosis of anaplastic oligodendroglioma. Anaplastic oligodendrogliomas (WHO grade III) are therefore defined by the presence of brisk mitotic activity and/or endothelial hypertrophy. Currently, a WHO grade IV is not recognized for oligodendroglial tumors, and among diffuse gliomas it is restricted only to glioblastoma, an astrocytic neoplasm by definition. The diagnosis of diffuse gliomas with mixed features (i.e., oligoastrocytomas) has gradually fallen out of favor, since there is no distinct molecular signature for these tumors [2, 3]. The caveat is that truly mixed gliomas at the morphologic and molecular level exceptionally occur [4, 5].
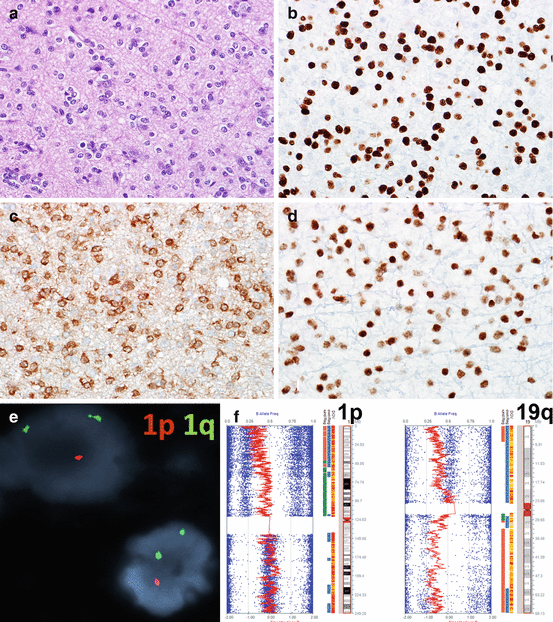
Fig. 5.3
Biomarkers in oligodendrogliomas. Oligodendrogliomas are characterized at the histologic level by the presence of round, uniform cells with little pleomorphism and frequent perinuclear halos (a). OLIG2 expression is a feature of almost all oligodendrogliomas (b), but may also be present in other glioma subtypes. Mutant IDH1 (R132H) protein expression (c) and preserved ATRX expression (d) are additional phenotypic features. The most relevant test for oligodendrogliomas is 1p19q, and the presence of 1p19q co-deletion represents a diagnostic, prognostic, and predictive biomarker in these tumors. This may be identified by FISH (1p loss illustrated) (e) or SNP array (f). The main advantage of the latter is that it allows the identification of whole-arm 1p and 19q loss, as reflected by the leftward shift on the red LRR line (Color figure online)
Historically, histologic subtype and grade were the cornerstone of prognostication in the evaluation of diffuse gliomas. However, with the gradual identification of robust diagnostic, prognostic, and predictive biomarkers, increasing precision has been achieved in the classification of these tumors. For instance, the combination of just three biomarkers (1p19q co-deletion, TERT promoter mutation, and IDH1 or 2 mutations) has been proposed as a robust molecular-based classification for gliomas in a recent large multi-institutional study [6].
Molecular Subclasses of Glioblastoma
Gene expression profiling is a robust tool that has facilitated the identification of molecular classification of high-grade gliomas, particularly glioblastoma [7]. In a hallmark study, Phillips et al. identified molecular subclasses of high-grade gliomas, predominantly glioblastomas that have been used as a basis for several molecular classification efforts [8]. By k-means clustering, three different subclasses were identified in a set of 76 high-grade gliomas (WHO grade III and IV), termed proneural, mesenchymal, and proliferative, based on the dominant pattern of gene overexpression. The molecular subclasses were subsequently validated in an independent set of glioblastomas. The overall finding of this study, of relevance to future profiling efforts, is that the molecular subclasses had a more powerful prognostic value, compared to classic histologic grading schemes.
In a subsequent analysis capitalizing on the wealth of multidimensional molecular data on glioblastoma provided by TCGA, Verhaak et al. [9] described four molecular classes of glioblastoma based on gene expression profiling, but also containing distinctive genetic alterations. These classes included the “classic” (high-level EGFR amplification, Ch10 loss), “mesenchymal” (NF1 gene alterations), “proneural” (PDGFRA gains, IDH1 and TP53 mutations), and a “neural” subtype characterized by the expression of neuronal genes, as the name implies. Of clinical relevance, the proneural group had a trend for better outcome. However, aggressive chemoradiation treatments appeared to benefit classical and mesenchymal subgroups better.
An additional improvement in molecular classification in neoplastic neuropathology has been the implementation of high-resolution methylation arrays. Again, using TCGA data, Noushmehr et al. described a CpG island methylator phenotype (CIMP) in glioblastoma [10]. The CIMP phenotype was initially described in colon cancer, and subsequently identified in other cancer types, defined by the presence of CpG island methylation in a subset of genes of a subset of tumors. The CIMP phenotype in glioblastoma is associated with the proneural molecular subgroup and IDH mutations and is more frequent in younger patients, lower-grade gliomas, and better clinical outcome. In fact, this CIMP phenotype may be a consequence of IDH mutations [11]. Applying methylation profiling with the Illumina 450k methylation array to adult and pediatric glioblastoma datasets, Sturm D et al. reported six distinct epigenetic subsets of glioblastoma enriched for specific mutations (e.g., IDH1, H3F3A G34, H3F3A K27) and associated with distinct clinical features (e.g., age, anatomic location, outcome).
1p19q Co-deletion
One of the first and most robust biomarkers in neuro-oncology is 1p19q co-deletion. Early cytogenetic observations uncovered a high frequency of 1p and/or 19q co-deletion in the subset of adult diffuse gliomas with oligodendroglial morphology [12, 13]. Subsequent retrospective studies confirmed that the chemosensitivity of oligodendroglial tumors was related to this specific genetic alteration [14]. These findings were subsequently confirmed by two independent clinical trials. The mechanisms underlying this peculiar cytogenetic alteration is an unbalanced t(1;19) translocation [15, 16]. Subsequent whole-exome sequencing studies identified mutations in the putative tumor suppressor genes FUBP1 (Chr 1p) and CIC (Chr 19q) in many, but not all, tumors with 1p19q co-deletion [17, 18]. Oligodendrogliomas with 1p19q co-deletion also have frequent IDH1 or 2 mutations and TERT promoter mutations (Fig. 5.3) [2].
Currently, 1p19q testing is recommended for all oligodendroglial tumors given its strong prognostic impact and at the current time provides a rationale for PCV chemotherapy, given the positive effect on overall survival on this molecular subset of tumors in the RTOG 9402 and EORTC 26951 trials [19, 20]. Molecular testing for 1p19q co-deletion is performed in most laboratories in which primary brain tumors are frequently evaluated. Since 1p19q co-deletions are large, involving essentially the whole arms, it may be tested by a variety of molecular techniques, most commonly FISH, SNP/CGH arrays, and PCR-based microsatellite analysis. All these techniques have their advantages and disadvantages. FISH testing requires only two tissue slides and may identify the abnormality even when present focally in the tissue. Conversely, SNP/CGH arrays are able to separate whole-arm deletions, which are most specific for oligodendroglial tumors, from partial/focal chromosomal deletions that may be identified in a variety of tumor types.
IDH1–2 Mutations
Mutations in IDH1 or 2 are relatively uncommon in cancer, but a frequent genetic alteration of diffuse gliomas grade II and III, astrocytic and oligodendroglial, as well as secondary glioblastomas. The IDH1 and IDH2 genes encode for NADP+-dependent enzymes involved in the metabolism of citrate, with IDH1 residing in the cytosol and IDH2 in mitochondria. Given that mutations are almost always confined to the same codon, early suspicions were that the mutations were gain of function. Currently, it is known that mutant forms of IDH1 and 2 lead to the production of an abnormal metabolite 2-hydroxyglutarate (2-HG) [21]. Testing for these mutations is relatively straightforward at the present time. An antibody applicable to immunohistochemistry has been developed against the most frequent mutant protein, i.e. IDH1 (R132H) [22, 23]. The remaining mutations may be identified by PCR-based techniques and DNA sequencing. More recently, inhibitors of IDH1 have been developed, and the identification of these mutations is a prerequisite for clinical trials involving this compound.
TERT Promoter Mutations, ATRX Mutations, and the Alternative Lengthening of Telomere (ALT) Phenotype
Telomere maintenance is an essential requirement for cell survival, including cancer cells. In diffuse gliomas, this occurs through two different mechanisms, by increasing telomerase expression or through the alternative lengthening of telomeres (ALT). Genetic alterations associated with these two phenotypes are TERT promoter mutations (activating) [24] and ATRX mutations (inactivating) [25]. These genetic alterations are mutually exclusive most of the time and generally recognize tumors with different molecular signatures. However, on rare instances, they may coexist [6].
Sequencing techniques are ideally suited to identify TERT promoter mutations, since they are activating and therefore are almost always of one of two types (C228T or C250T). Conversely, ATRX mutations are inactivating and occur through multiple sites in this relatively large gene. Therefore, immunohistochemistry demonstrating absence of the protein product, with relative preservation in the nonneoplastic underlying cell components, is more feasible. ATRX is one of several chromatin remodeling proteins, and its loss in neoplastic cells is tightly correlated with the ALT phenotype. The latter may be identified using telomere-specific FISH, which demonstrates diagnostic ultrabright signals [26].
MGMT Promoter Methylation
Esteller et al. reported an association of methylation of the gene promoter of MGMT, which encodes for a DNA repair enzyme, and response of high-grade astrocytomas to alkylating agents [27]. This early observation was confirmed in tumor tissue obtained from a subsequent clinical trial using temozolomide in newly diagnosed glioblastoma [28]. Currently, testing for MGMT promoter methylation has become standard of molecular testing for high-grade astrocytomas, where it is considered a predictive marker for response to temozolomide. In addition, MGMT methylation is associated with the phenomenon of “pseudoprogression,” where early changes on imaging scans after treatment are related to treatment effects rather than tumor recurrence [29]. However, MGMT methylation does not necessarily dictate treatment, since a subset of unmethylated tumors by commonly used techniques also benefit from temozolomide regimens. However, MGMT methylation status is a prerequisite for enrollment in many clinical trials, which provides a rationale for testing it routinely.
One specific problem encountered with molecular testing for MGMT methylation is that the gene promoter contains many CpG islands, and only a small region of the promoter is studied as a surrogate to interpret the gene as methylated. Although conceptually it would make sense to test for the MGMT protein as a surrogate of methylation/gene inactivation, immunohistochemistry may not be useful in the identification of the favorable high-grade astrocytoma subset associated with methylation [30].
EGFR Amplification and PTEN Loss
EGFR is frequently activated in high-grade astrocytomas, particularly glioblastomas. The most relevant mechanisms include receptor amplification, as well as the constitutively active EGFRvIII mutation which lacks the extracellular domain. Early preclinical data suggested that glioblastomas with EGFR alterations could benefit from inhibitors, and a retrospective study provided data supporting that the subset of tumors with intact PTEN expression was the most likely to benefit. Unfortunately, EGFR inhibition has not proven efficacious in some clinical trials, and therefore EGFR status is not tested routinely in all laboratories. However, the presence of EGFRvIII may qualify patients to clinical trials using specific antibodies as a therapeutic strategy against tumors with this specific alteration. The most frequent method to test for EGFR amplification is FISH, while EGFRvIII may be identified by PCR-based methods or immunohistochemistry.
Pediatric Gliomas
Primary CNS tumors are the most common solid tumors and the leading cause of cancer-related death in children. More than 4000 pediatric CNS tumors are diagnosed each year in the United States (cancer.org). Among pediatric CNS tumors, gliomas are the most common type, accounting for approximately 60 % of the cases [31]. Pediatric gliomas are a heterogeneous group of neoplasms and are clinically, histologically, and molecularly distinct from their adult counterparts [32]. Unlike the adult population among which high-grade gliomas (HGGs) represent the predominant type of CNS tumors, low-grade gliomas (LGGs) are far more common in children. Over the last few decades, a wealth of data has emerged from basic and translational research of pediatric gliomas, which helps to elucidate the biologic and molecular makeup of these tumors [33]. In particular, the advances of next-generation sequencing technologies have resulted in a remarkable progress in the characterization of key genetic alterations in both low-grade and high-grade pediatric gliomas. The data not only provide a better understanding of tumor biology but also highlight the potential for development of novel treatment strategies [33]. In addition to their diagnostic use, the molecular markers of tumors offer useful information for prognostic risk stratification and customized treatment planning.
Molecular findings of each pathologic entity of pediatric gliomas are described as follows.
Pilocytic Astrocytoma and the Variants
Pilocytic astrocytomas (PAs) are the most common brain tumors of childhood. The incidence rate in the United States is 0.84 per 100,000 [1]. PAs most frequently occur in the cerebellum, followed by hypothalamus/optic pathway, brainstem, and spinal cord. PAs in general are considered benign tumors. The vast majority of the cases are WHO grade I and rarely progress to high-grade tumors. When completely resected, most cases have a favorable outcome, with a 10-year survival of over 90 % [34].
NF1-Associated Tumors
PA arises in 15–20 % of individuals with neurofibromatosis type 1 (NF1). It can affect NF1 patients at any age [35]. Since most NF1-associated PAs occur at anatomic sites along the visual pathways, the tumors are also called “optic pathway gliomas.” Because the natural history and treatment of NF1-associated tumors are different from that of sporadic PAs [35], it is important to identify tumors associated with this particular genetic condition. Due to the indolent nature of the NF1-associated PAs, the tumors are rarely resected or even biopsied.
NF1 is one of the largest human genes, composed of 60 exons and spanning more than 300 kb of genomic DNA. Mutation detection in NF1 is complicated by the gene’s large size, the lack of mutation hotspots, the presence of pseudogenes, and the wide variety of possible genetic alterations [36]. Due to the mutational heterogeneity, comprehensive genetic evaluation is recommended for NF1 patients. The testing generally includes sequencing of all coding exons, copy number analysis, and assessment of deep intronic splice mutations through RNA studies [37]. Despite the fact that multiple unique mutations are found throughout the entire gene, there is no clear association between specific mutations and clinical phenotypes [38].
The NF1-encoded protein neurofibromin is a negative regulator of RAS, a small GTPase in the mitogen-activated protein kinase (MAPK) pathway (Fig. 5.4) [39]. This finding, along with the discovery of frequent genetic alterations of MAPK pathway components in sporadic tumors, indicates that PAs represent a single-pathway disease [34].
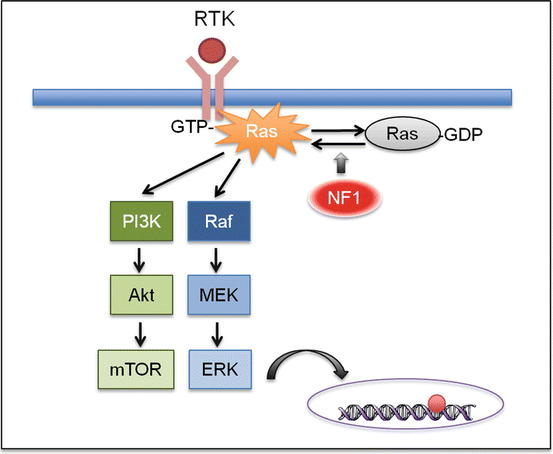
Fig. 5.4
NF1 signaling. NF1 encodes for neurofibromin, a negative regulator of RAS, a small GTPase that transduces signal from the receptor tyrosine kinase (RTK). Therefore, loss of NF1 expression leads to elevated RAS activity. The subsequent activation of the MAPK and mTOR signaling leads to cell proliferation and tumorigenesis
BRAF Fusion Tumors: Pilocytic Astrocytoma (PA), Pilomyxoid Astrocytoma (PMA), and Disseminated Oligodendroglial-Like Leptomeningeal Neoplasm (DOLN)
The most common genetic alteration of non-NF1-associated PAs is a tandem duplication of ~2 Mb at chromosome 7q34, resulting in a gene fusion between the N-terminus of KIAA1549 and the kinase domain of BRAF (Fig. 5.5) [34, 40]. BRAF, a serine/threonine kinase, is a downstream effector of RAS in the MAPK pathway. The most common fusion is between exon 16 of KIAA1549 and exon 9 of BRAF, followed by 15-9 and 16-11 [40, 41]. The N-terminal end of the KIAA1549 protein replaces the auto-inhibitory domain of BRAF and therefore constitutively activates the kinase and the downstream MAPK signaling [40]. This rearrangement event is detected in ~70 % of PAs [40, 42]: much more frequent in the posterior fossa tumors (~90 %) than other anatomic locations [34, 41, 43]. Besides KIAA1549, several other genes including FAM131B, RNF130, CLCN6, MKRN1, GNA11, QKI, FZR1, and MACF1 have been identified as fusion partners for BRAF but at much lower frequencies [34, 41, 43, 44].
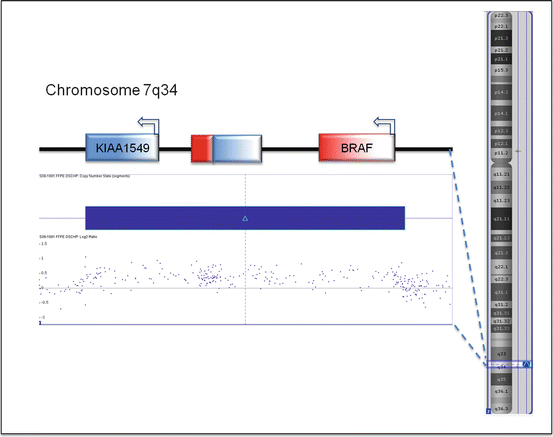
Fig. 5.5
KIAA1549–BRAF fusion. The fusion results from tandem duplication of a 2 MB segment of chromosome 7q34. It combines the N-terminal half of KIAA1549 and the kinase domain in the C-terminal half of BRAF. The tandem duplication can be detected by SNP array
In addition to BRAF fusion, genetic alterations involving genes such as FGFR1, NTRK, RAF1, and KRAS have recently been reported in a small number of PAs, especially the non-cerebellar tumors. Various aberrations in FGFR1 have been identified, including internal tandem duplication (FGFR1-ITD), FGFR1–TACC1 fusion, and point mutations in its tyrosine kinase domains. The genetic alterations in NTRK and RAF1, such as NAV1–NTRK2, QKI–NTRK2, NACC2–NTRK2, BRAF–RAF1, QKI–RAF1, and SRGAP3–RAF1, are exclusively gene fusions. These causative genes apparently all belong to the MAPK pathway, and the alterations presumably cause activation of the MAPK signaling. In the appropriate clinicopathological context, the presence of these genetic alterations, particularly KIAA1549–BRAF fusion, strongly supports a diagnosis of PA. The KIAA1549–BRAF fusion can be detected by FISH, SNP array, PCR, or next-generation sequencing technology.
Pilomyxoid astrocytoma (PMA) is a variant of PA that arises from the hypothalamic region and affects children under age 2 [45]. It may have frequent recurrences and more aggressive clinical behavior. Unlike classic PAs which demonstrate a biphasic architecture and harbor frequent Rosenthal fibers and eosinophilic granular bodies, PMAs are predominantly monophasic, consisting mainly of small bipolar cells in a myxoid matrix.
While morphologically dissimilar, PAs and PMAs are often considered two ends of the spectrum. Some PMAs display a “maturational effect” during the disease process, becoming more PA-like over time. These tumors are termed “intermediate pilomyxoid tumors” [46]. In terms of the genetic makeup, PMAs and intermediate pilomyxoid tumors are similar to PAs, with the majority of the cases carrying the KIAA1549:BRAF fusion. However, there appears to be a significant portion of PMAs that are negative for BRAF alterations [47]. It is unclear whether the BRAF wild-type tumors represent distinct molecular entities.
There is another distinct pediatric CNS tumor known to carry KIAA1549:BRAF gene fusion: disseminated oligodendroglial-like leptomeningeal neoplasm (DOLN) [48, 49]. In addition to BRAF fusion, the tumor concurrently demonstrates loss of chromosome 1p [49]. The tumor has a tendency for extensive dissemination and may demonstrate an indolent behavior despite the presence of widespread dissemination, a key characteristic of this tumor type [48].
BRAF V600-Mutant Tumors: Pleomorphic Xanthoastrocytoma (PXA), Ganglioglioma, Diencephalic Pediatric Low-Grade Astrocytoma (PLGA), Dysembryoplastic Neuroepithelial Tumor (DNT), Epithelioid Glioblastoma, and Pediatric Secondary High-Grade Glioma (sHGG)
The most common BRAF mutation is the c.1799T>A point mutation, which results in a valine (V) to glutamic acid (E) substitution at position 600. The mutation causes constitutive activation of the kinase, resulting in a 500-fold increase in its activity [50]. Other BRAF kinase-activating mutations have been reported, but at substantially lower frequencies. They are mainly in the form of point mutations and located in the kinase domain.
BRAF V600E mutation is frequently associated with melanoma, colorectal cancer, and thyroid cancer [51]. Several molecular platforms have been utilized to detect BRAF point mutations in tumor samples. These include Sanger sequencing, pyrosequencing, allele-specific real-time polymerase chain reaction (PCR), high-resolution melting curve analysis, and next-generation sequencing methods [52]. Currently, the Food and Drug Administration (FDA) has approved two molecular platforms as companion diagnostic tests for BRAF or MEK inhibitors. Both tests are designed specifically for detection of the V600E mutation in FFPE melanoma samples. The COBAS 4800 BRAF V600 Mutation Test developed by Roche Molecular Systems (Basel, Switzerland) is a companion test for the BRAF inhibitor vemurafenib, whereas the THxID™ BRAF Kit from bioMérieux Inc. (Marcy-l’Étoile, France) is a test for the MEK inhibitor trametinib and BRAF inhibitor dabrafenib. The analytic performance of these PCR-based assays has demonstrated >99 % sensitivity in the detection of BRAF V600E mutation. The lower limit of detection of the mutant alleles is reportedly less than 4–5 % [52]. In addition to molecular techniques, a monoclonal antibody VE1 was recently developed to detect BRAF V600E mutant protein. It has been successfully used in various types of tumors, including pleomorphic xanthoastrocytoma (PXAs) [53].
Among CNS tumors, BRAF V600 mutations are most prevalent in pediatric gliomas and glioneuronal tumors. Approximately 60–70 % of pleomorphic xanthoastrocytomas (PXAs) and 20–60 % of gangliogliomas harbor BRAF V600E mutation [54, 55]. Additionally, the mutation is detected in 30–50 % of dysembryoplastic neuroepithelial tumors (DNTs) [56], diencephalic pediatric low-grade astrocytomas (PLGAs) [47], pediatric secondary high-grade gliomas (sHGGs) [57], and epithelioid glioblastoma [58]. A significant portion of BRAF V600E PXAs and sHGGs concurrently demonstrate CDKN2A deletion [41, 57]. CDKN2A is an important tumor suppressor gene, the deletion of which leads to genomic instability [59].
Given the high frequency of BRAF alterations among pediatric gliomas, there is considerable interest in targeted inhibition of the MAPK pathway as a new treatment strategy. BRAF or MEK inhibitors, such as sorafenib, selumetinib (AZD6244), and vemurafenib, are currently being investigated in patients with BRAF-mutant low-grade gliomas, especially those with recurrent or refractory disease [60].
Cerebral Hemispheric Diffuse Low-Grade Gliomas: Diffuse Astrocytoma, Angiocentric Glioma, and Oligodendroglioma
Diffuse PLGAs comprise several molecularly distinct entities. Unlike adult infiltrating gliomas, diffuse PLGAs infrequently harbor the IDH1/2 mutation [61, 62]. In contrast, genetic alterations of FGFR1 or the transcription factor MYB/MYBL1 are commonly reported in WHO grade II diffuse astrocytomas, each accounting for ~30 % of the cases [41, 63]. The FGFR1 aberration in diffuse astrocytomas is the same tandem duplication observed in rare cases of PAs [41]. The MYB/MYBL1 rearrangements result in partial deletion of MYB or partial duplication of MYBL1 with truncation of its C-terminal negative-regulatory domain. A similar MYB deletion-truncation is identified in angiocentric gliomas, a WHO grade I glioma [63]. It remains unclear whether these novel alterations correlate with an indolent behavior or a tendency toward progression. Their prognostic significance awaits further investigation.
Oligodendrogliomas are a rare tumor of childhood. In adults, co-deletion of chromosome whole arms 1p and 19q together with IDH1 point mutation has become a molecular hallmark of oligodendroglioma and correlates with a favorable clinical outcome [2]. Adult oligodendrogliomas also frequently harbor mutations in CIC, FUBP1, NOTCH1, and the TERT promoter [2, 17]. Pediatric oligodendrogliomas, on the other hand, lack these genetic alterations. 1p/19q co-deletion appears infrequent in pediatric compared to adult oligodendrogliomas and are virtually absent in tumors presenting in the first decade of life [64]. Despite frequent chromosomal copy number variations, no consistent recurrent genetic abnormalities have been identified in pediatric oligodendrogliomas [65].
Pediatric oligodendrogliomas are predominantly low grade. Clinically, they appear more indolent than their adult counterpart, demonstrating a low rate of histologic progression [66]. Histologically, at least a subset of pediatric oligodendrogliomas resembles DNT, a lesion whose relationship to oligodendroglioma remains to be defined. However, the higher propensity for recurrence, the presence of anaplastic examples, and the presence of variable cytogenetic alterations in pediatric oligodendroglioma suggests that they represent different entities [65].
Diffuse Intrinsic Pontine Glioma (DIPG)
Diffuse intrinsic pontine glioma (DIPG) is an infiltrative glial neoplasm arising from the brainstem. Due to the poor surgical resectability and aggressive growth, the tumor has a dismal prognosis. The median survival for children with DIPG is less than 1 year from diagnosis, and no improvement in survival has been realized in more than three decades [67]. In recent years, advances in genetics and molecular biology have significantly improved our knowledge of DIPGs. Although DIPGs are histologically consistent with fibrillary astrocytomas, recent data indicated that DIPGs have a different genetic makeup than most supratentorial infiltrating gliomas.
Based on the molecular features, DIPG can be divided into three subgroups: H3-K27M, Silent, and MYCN [68]. H3-K27M DIPGs are the predominant subgroup, accounting for ~80 % of the cases. Seventy to eighty percent H3-K27M DIPGs carry a key driver mutation in H3F3A, which encodes the replication-independent histone 3 variant H3.3 [69–71]. The rest of the K27M tumors have the mutation in the HIST1H3B gene, which encodes the histone variant H3.1 [70]. Histone is a main component of chromatin. Its main function is to wrap DNA into small units called nucleosomes. The fact that the mutations invariably occur at Lys27, a site of key regulatory posttranslational modifications, suggests that DIPGs may be driven by altered epigenetic states. The K27M mutation was found to cause a global reduction of H3K27 trimethylation (H3K27me3) by inhibiting the enzymatic activity of the polycomb repressive complex 2 (PRC2). This K-to-M substitution may represent a mechanism of epigenetic alteration leading to tumorigenesis [72].
The H3-K27M subgroup tumors commonly harbor other genetic alterations, such as TP53 mutation, ACVR1 (activin A receptor, type I) mutation, and PDGFRA (platelet-derived growth factor receptor α) amplification. The tumors positive for both H3-K27M and TP53 mutations have highly unstable genomes and often show high-grade histology [73]. ACVR1 is a member of the bone morphogenic protein (BMP) signaling pathway. Its mutation is detected in 20–30 % of DIPGs, particularly those with H3.1 mutation [68, 74, 75]. PDGFRα is a receptor tyrosine kinase. The finding of PDGFRA amplification in 40 % of H3.3 mutant DIPGs [69] highlights the potential of tyrosine kinase inhibition as a novel therapeutic strategy for this subgroup of tumors.
Compared to the H3-K27M tumors, the MYCN subgroup has no recurrent mutations but is instead characterized by hypermethylation, high-grade histology, and MYCN amplification. The Silent subgroup has stable genomes and a low-grade histology. Strangely, there is no difference in overall survival of the Silent subgroup in comparison with the other two subgroups [68].
Supratentorial High-Grade Glioma
Pediatric supratentorial HGGs are highly heterogeneous and aggressive brain tumors. Recent studies have highlighted crucial molecular differences between these tumors and their adult counterparts [33]. For instance, pediatric HGGs appear more genomically stable, demonstrating less copy number variation than adult HGGs [76]. Pediatric HGGs also lack EGFR (epidermal growth factor receptor) amplification, a phenomenon commonly observed in adult glioblastomas [77].
Recent genomic studies have identified several novel genetic aberrations in pediatric supratentorial HGGs. These genetic alterations are extremely rare in adult HGGs.
Ten to twenty percent of the pediatric supratentorial HGGs have Gly34Arg (G34R) or Gly34Val (G34V) substitution in histone H3.3 [69]. These mutations are predominantly found in older children and young adults [70]. The G34R/V-H3.3 mutant tumors invariably carry mutations in TP53 and chromatin remodeling genes ATRX (α-thalassemia/mental retardation syndrome X-linked) or DAXX (death-domain-associated protein). The H3F3A/TP53/ATRX–DAXX mutant tumors have specific gene expression profiles and demonstrate alternative lengthening of telomeres [70].
Besides G34R/V-H3.3 mutations, mutations in SETD2, a H3K36 trimethyltransferase, are identified in 15 % of pediatric HGGs. SETD2 mutations occur in older children and young adults and are specific to HGG of the cerebral cortex, similar to the H3.3 G34R/V and IDH1/2 mutations. Most SETD2 alterations are truncating mutations. They are mutually exclusive with H3F3A mutations but partly overlapped with IDH1 mutations. SETD2-mutant tumors showed a substantial decrease in H3K36me3 levels, indicating that the mutations are loss of function [78].
Embryonal Tumors
CNS embryonal tumors represent a heterogeneous group of highly malignant neoplasms, all of which are WHO grade IV. Their primitive, poorly differentiated appearance morphologically resembles neural stem cells. These tumors have a predilection for young children. They not only invade nervous tissue, but can also disseminate into the cerebrospinal fluid (CSF) and subarachnoid space [79].
This section will discuss major entities of embryonal tumors, including medulloblastoma, pineoblastoma, embryonal tumor with multilayered rosettes (ETMR), and atypical teratoid/rhabdoid tumor (AT/RT). Except for AT/RT, the rest of the tumors are all considered poorly differentiated tumors of neuronal lineage.
Medulloblastoma
Under the current consensus classification, medulloblastomas are divided into four principle molecular subgroups: WNT, Sonic Hedgehog (SHH), group 3, and group 4 (Fig. 5.6). The subgrouping is based on differences in the gene expression profiles. SHH, group 3, and group 4 each contributes approximately one-quarter to one-third to the medulloblastomas, whereas the WNT subgroup accounts for 10–15 % of the cases [80, 81]. Each subgroup may be further subclassified into more than one subset. While the subsets of the subgroups seem readily apparent, they are not well characterized at this point [82, 83].
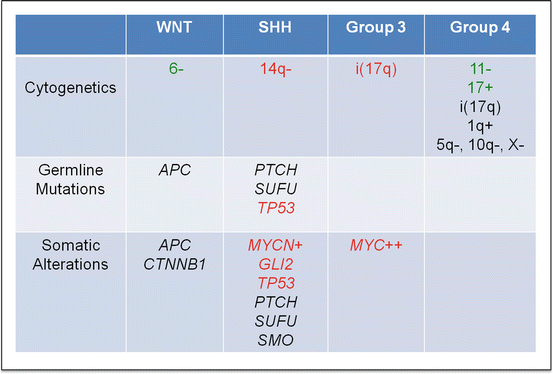
Fig. 5.6
Important cytogenetic and molecular findings in four subgroups of medulloblastoma. Good prognostic predictors are highlighted in green, and poor prognostic predictors are highlighted in red (Color figure online)
A recent study indicated subgroup and metastatic status as the two most powerful predictive prognostic biomarkers for medulloblastomas [84], highlighting the importance of including molecular subgrouping in initial workup of tumors. Molecular subgrouping is not only useful in the setting of risk stratification for a prospective clinical trial but also crucial for patient selection for targeted therapy (e.g., SHH inhibitor).
Molecular subgrouping of medulloblastomas can be achieved by immunohistochemistry (IHC) [80, 82], gene expression profiling [85], or DNA methylation profiling [81]. IHC studies utilize antibodies against four markers: GAB1, β-catenin, filamin A, and YAP1. Immunoreactivity for GAB1 characterizes SHH tumors, and nuclear staining for β-catenin identifies WNT tumors. SHH and WNT tumors both show immunoreactivity for filamin A and YAP1. IHC has been shown to reliably classify ~98 % medulloblastoma FFPE samples. Its fast turnaround time and low costs are also appealing points. However, issues such as lot-to-lot variability of antibodies, interinstitutional differences in tissue fixation, and embedding, technical variations, and inter- and intraobserver variability in image interpretation remain problematic [85]. In addition, IHC cannot distinguish between group 3 and group 4 tumors (both designated as non-SHH/WNT subgroup by IHC analysis) [80].
To develop a rapid, reliable, and reproducible method for medulloblastoma subgrouping, Northcott and colleagues have taken advantage of the nanoString nCounter System, a nonenzymatic multiplexed assay that uses sequence-specific probes to digitally measure target abundance (i.e., mRNA) within a given sample. They designed a custom CodeSet (i.e., probe library) consisting of interrogating probes against 22 medulloblastoma subgroup-specific signature genes. There are 5–6 signature genes included for subgroup WNT: WIF1, TNC, GAD1, DKK2, EMX2; SHH: PDLIM3, EYA1, HHIP, ATOH1, SFRP1; group 3: IMPG2, GABRA5, EGFL11, NRL, MAB21L2, NPR3; and group 4: KCNA1, EOMES, KHDRBS2, RBM24, UNC5D, OAS1. Northcott et al. have shown that the nanoString assay confidently predicts subgroup in 88 % of recent (<8 years) FFPE cases, of which 100 % have accurate subgroup assignment. The rapid turnaround and high reproducibility also make it highly suited for future medulloblastoma clinical trials [85].
Medulloblastomas can also be subclassified based on their DNA methylation profiling [81]. The concept of molecular subgrouping by methylation patterns is very similar to that of gene expression profiling. One additional advantage methylation profiling offers is the capability of detecting copy number alterations, such as monosomy 6 and MYC amplification. The sensitivity of high-density DNA methylation arrays, such as Infinium HumanMethylation450 BeadChips, in detecting copy number variations is comparable to most SNP platforms [86].
The distinctive characteristics of the four medulloblastoma subgroups are described in detail below.
WNT Subgroup
WNT tumors have the most favorable prognosis among all four medulloblastoma subgroups. They can occur at all ages, but are uncommon in infants. WNT medulloblastomas have a known association with Turcot syndrome, a cancer syndrome caused by germline mutations in APC (adenomatous polyposis coli). APC plays an important role in the WNT signaling pathway by forming a complex with β-catenin and preventing its translocation to the nucleus (Fig. 5.7). In addition to germline APC mutations, somatic APC and CTNNB1 (gene encoding β-catenin) mutations have also been found in sporadic medulloblastomas [87, 88]. These germline and somatic genetic data strongly support an etiological role for canonical WNT signaling in the pathogenesis of this group of tumors [83].
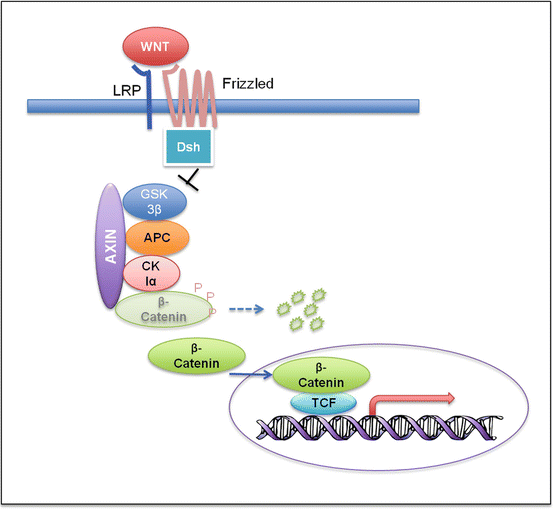
Fig. 5.7
Wnt signaling pathway. In the absence of the Wnt ligand, ß-catenin forms a complex with Axin, APC, GSK3ß, and CK1α, becomes phosphorylated, and gets degraded through the ubiquitin proteasome pathway. Binding of Wnt to the Frizzled/LRP receptor complex causes hypophosphorylation of ß-catenin through inhibition of GSK3ß by Dishevelled (Dsh). Hypophosphorylation stabilizes ß-catenin, which accumulates in the cytoplasm and translocates to the nucleus. It then activates the transcription of the target genes with the transcription factor TCF
The majority of the WNT medulloblastomas belong to the classic histologic subtype. Large cell/anaplastic (LCA) histology is uncommon. WNT medulloblastomas can be identified by nuclear immunoreactivity for β-catenin. In cases that have equivocal IHC results, evaluation for CTNNB1 mutation or monosomy 6 can sometimes help to confirm the diagnosis [80]. Nevertheless, using monosomy 6 as the lone diagnostic criterion for WNT medulloblastomas can be problematic, since the finding is observed in 14 % non-WNT tumors and absent in 21 % WNT tumors.
Medulloblastomas with monosomy 6 are associated with an excellent prognosis. According to Shih et al., the prognostic significance of monosomy 6 is driven by its enrichment in WNT subgroup, and it thus holds no further significance in subgroup-specific analysis [84]. Similarly, somatic TP53 mutations observed in 16 % of WNT medulloblastomas are of no significant prognostic value [89].
Sonic Hedgehog (SHH) Subgroup
Medulloblastomas of the nodular/desmoplastic subtype are known to have a strong association with the SHH subgroup. However, morphology alone is not a reliable marker as up to 50 % of SHH medulloblastomas can demonstrate classic or LCA histology [83]. While identification of SHH tumors can be performed by IHC (demonstrating immunoreactivity for GAB1, filamin A and YAP1) [80], the method fails to detect a small fraction of tumors that do not express the SHH markers. Therefore, transcriptional profiling remains a robust modality to diagnose SHH medulloblastomas.
Overall, the prognosis of SHH medulloblastomas is intermediate between that of WNT (good) and group 3 tumors (poor). SHH tumors demonstrate a unique bimodal age distribution, with two peak incidences in infants (≤3 years) and adults (≥18 years). Recent data have indicated that SHH medulloblastomas in infants, children, and adults are three distinct molecular and risk groups [84, 90]. Childhood (age 4–17) SHH tumors, although not as common, are associated with genomic instability and a poor survival. Tumors in this age group frequently harbor TP53 mutations (~50 % germline) and amplification of GLI2 and MYCN [89, 90]. Besides GLI2 and MYCN amplification, loss of chromosome 14q confers significantly inferior survival. Shih et al. propose using two molecular biomarkers (GLI2 and 14q FISH) in conjunction with the metastatic status to stratify patients with SHH medulloblastoma [84].
SHH pathway (Fig. 5.8) mutations have been found in both germline and somatic form. Germline mutations include mutations in the SHH receptor PTCH (Gorlin syndrome) and SHH inhibitor SUFU. Similarly, somatic SHH pathway mutations in PTCH, SMO, or SUFU have been frequently reported in sporadic medulloblastomas [83]. SUFU mutations are restricted to infants, whereas SMO mutations are enriched in adults. All these mutations are mutually exclusive [90].
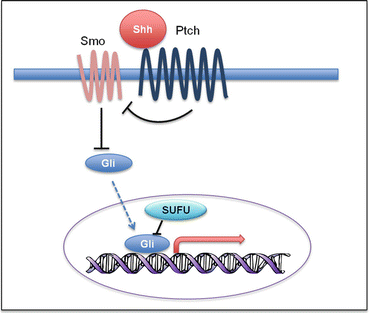
Fig. 5.8
Shh signaling pathway. Shh pathway activation is initiated by binding of Shh to the receptor Ptch. This relieves the inhibition of Gli by Smoothened (Smo). Gli subsequently translocates to the nucleus and activates transcription of Shh target genes
SHH pathway antagonists, primarily those inhibiting at the level of smoothened (SMO), are currently being investigated as a novel targeted therapeutic agent for SHH medulloblastomas [90]. Kool and colleagues have observed in xenograft mouse models that only SHH tumors harboring a PTCH1 mutation are responsive to SMO inhibition. Tumors harboring an SUFU mutation or MYCN amplification are resistant to the therapy. Therefore, assessment of the PTCH1 mutation status is important for guiding SHH pathway-targeted therapies.
Group 3
Group 3 medulloblastomas occur in infants and children, but are rare in adults. The current gold standard for diagnosis of a group 3 tumor is transcriptional profiling. Group 3 medulloblastomas can be further subclassified based on the metastasis, MYC amplification, and isochromosome 17q status. Patients with metastasis, iso17q, or MYC amplification represent the high-risk group [84]. MYC-amplified tumors also have a high incidence of LCA histology [83]. Nonmetastatic group 3 tumors negative for iso17q and MYC amplification have a clinical outcome similar to SHH and group 4 tumors [83, 84].
Group 4
Group 4 medulloblastomas, being the greatest in number, are least characterized compared to the other subgroups. Tumors in this subgroup have an overall intermediate prognosis. Within group 4, tumors with chromosome 10p loss along with monosomy 11 or trisomy 17 exhibit better survival. There is no cytogenetic marker associated with poor prognosis [84].
Based on current literature, medulloblastoma workup procedures, including imaging and histologic and molecular studies, are summarized in a schematic diagram (Fig. 5.9).
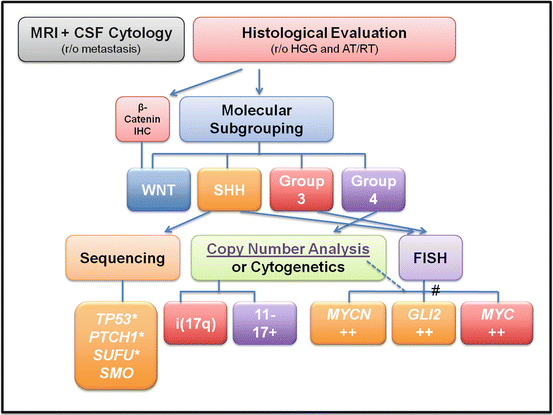
Fig. 5.9
Flowchart of the recommended diagnostic and prognostic workup for medulloblastoma. * Possible germline mutations. # Gene amplification can be detected by either FISH or high-resolution copy number analysis such as high-density SNP array
CNS Embryonal Neoplasms
A variety of embryonal neoplasms morphologically and immunophenotypically resembling medulloblastoma develop in the CNS. However, current understanding is that these tumors are clinically and molecularly distinct from medulloblastoma and represent a heterogeneous group. The term CNS-primitive neuroectodermal tumor (PNET) has been historically ascribed to these tumors, but this terminology has gradually fallen out of favor. Recent studies suggest the many embryonal neoplasms of the CNS comprise several well-defined entities, including atypical rhabdoid/teratoid tumor (AT/RT), embryonal tumor with multilayered rosettes (ETMR), HGG, and ependymoma, and should be reclassified based on IHC and molecular findings. For instance, embryonal tumors with loss of INI1 staining and SMARCB1 mutations/deletions should be considered AT/RT (Fig. 5.10). Cases without INI1 alterations are more complicated. Picard and colleagues suggest using cell lineage markers LIN28 and OLIG2 to classify CNS-embryonal tumors into three molecular groups: group 1 is primitive neural and positive for LIN28; group 2 is oligoneural and expresses OLIG2; and group 3 is the mesenchymal lineage that is negative for both LIN28 and OLIG2. Group 1 tumors also demonstrate frequent amplification of C19MC microRNA gene cluster [91, 92]. Clinically, patients with group 1 tumors are the youngest (< age 4) and have the poorest survival (median survival 0.8 years). Group 2 tumors have an intermediate clinical outcome, with a median survival of 1.8 years. Patients with group 3 tumors demonstrate the longest survival (median survival 4.3 years) but have the highest incidence of metastases at diagnosis [91].