Age of onset and progression of core clinical features of CADASIL.
Migraine
Migraine with aura is a common early symptom, affecting 40–60% of patients with CADASIL; the mean age of onset is 28.3 (± 11.7) years [17] but the onset of migraine has been reported in patients as young as 6 years [23]. The auras are predominantly visual and sensory, and most are indistinguishable from typical migraine with aura. However, there is a higher frequency of basilar, hemiplegic, and prolonged aura in this population. Hemiplegia may occur during a headache episode and the Notch3 gene is located in close proximity to the gene for familial hemiplegic migraine on chromosome 19 [14]. The frequency of attacks is variable and can range from one attack in a lifetime to several per month. Premenopausal women are more affected by migraine with aura than men or postmenopausal women [24]. Although white matter lesions are fairly common among patients with migraine, the pattern is usually milder in severity than in CADASIL [25], and the frequency of migraine with or without aura in patients with sporadic subcortical ischemic vascular disease is unclear. CADASIL provides an important window for exploring the mechanisms underlying the interface of migraine and cerebral ischemia.
Ischemic episodes
Ischemic stroke affects 85% of patients with CADASIL. It is the commonest initial presentation and onset can occur anytime between the third and sixth decade of life [13]. Ischemic stroke is more common in males compared to female CADASIL patients, contributing to higher Rankin scores and more severe executive dysfunction [24]. Clinically, patients present with classic lacunar syndromes that produce sensory or motor symptoms. However, some ischemic events will be clinically silent or produce mild and vague symptoms of dizziness, fatigue or confusion. Ischemic events usually recur, with an accumulation of deficits eventually leading to a classical stepwise decline, with gait difficulties, pseudobulbar palsy, urinary incontinence, and a dementia syndrome with frontal lobe features [26]. Ischemic events tend to have the greatest clinical impact in the fifties to seventies, with increasing evidence that disability in CADASIL is closely tied to the number and size of lacunar infarctions and hypodense lesions on T1-weighted MRI. Ischemic episodes usually occur earlier in CADASIL than in sporadic forms of subcortical vascular disease, with evidence of neurological resilience early in the course but classical stepwise deterioration late in the illness.
Psychiatric disturbance
Psychiatric disturbances are also evident in CADASIL, but behavioral symptoms are not well characterized. Families frequently note symptoms of irritability, mild depression, and a decline in motivation and speed of responding as the first signs of the illness [27, 28, 29]. Apathy is common in CADASIL patients and may contribute to a decrease in subjective quality of life [30]. Significant mood disturbance affects approximately 20% of patients [17, 27, 31]. Mood lability may manifest, with depression alternating with mania [13]. Pathological affect and disinhibition may occur in later stages. Panic attacks, schizophrenia, and personality changes have been reported [28, 29, 31, 32].
Cognitive impairment
Cognitive impairment generally occurs in the domains of attention, processing speed, executive functioning, and inefficient learning and retrieval of new information. Deficits are particularly evident on tasks requiring cognitive set-shifting, response inhibition, working memory, verbal fluency, and abstract concept formation [10, 33, 34, 35, 36]. Visuospatial impairments have been reported less frequently (e.g., [33, 34]). Phonemic verbal fluency was found to be consistently impaired in CADASIL, with greater levels of impairment than AD [11]. Episodic memory is generally well preserved until late in the illness [10] and tends to be characterized by deficits in encoding and retrieval rather than storage. Overall, the cognitive deficits elicited by formal neuropsychological assessment align well with the subjective complaints of patients in the early stages of the illness, which tend to focus on reduced mental efficiency, difficulties with multitasking, and poor recall. The cognitive deficits worsen with age [35] and with the presence of infarction [10], and demented and non-demented patients with CADASIL differ mainly by the severity rather than the pattern of cognitive deficits [36]. A review of some key studies of cognitive functioning in CADASIL follows.
Amberla et al. [10] found that patients with genetically confirmed CADASIL but no clinical evidence of transient ischemic attack, stroke or dementia performed more poorly than controls on tests of immediate memory, working memory, and executive functions. The authors interpreted this as evidence of incipient cognitive impairment in CADASIL even before the onset of clinical ischemic symptoms.
Peters et al. [36] reported on 65 patients with CADASIL and 30 control individuals matched for age, gender, and education. CADASIL subjects demonstrated pronounced deficits on measures of attention and psychomotor processing speed (e.g., Stroop interference condition, Trails B, and a composite score from performance on Symbol-Digit, digit-span backward, and digit cancellation tasks). The pattern of cognitive test performance was similar in CADASIL subjects who scored below the cut-off for dementia on the Mattis Dementia Rating Scale (i.e., ≤ 123 versus > 123) (n = 9) and those who scored above it (n = 56), but deficits were far more pronounced among the latter group. Moreover, compared to controls, CADASIL subjects over 45 years of age performed significantly more poorly on more tests than those under 45 years.
Peters et al. [37] also examined the 2-year progression of symptoms in 80 CADASIL individuals and demonstrated a non-significant trend toward decline on the Mattis Dementia Rating Scale. A significant correlation was found between increasing disability measured by the Rankin scale and scores on a structured interview for dementia. Peters et al. [36] found significant impairments in 65 mutation carriers compared to controls on tests of mental processing speed (e.g., Trail Making Test). Impairments of lesser extent were noted on measures of error monitoring (e.g., Stroop interference) and in verbal fluency and ideational praxis. Language functions, orientation, and recall were generally preserved.
Monet-Lepretre et al. [38] examined the impact of vascular risk factors in CADASIL compared to age-related subcortical ischemic vascular dementia (SIVD). CADASIL participants had higher body mass index than the SIVD group but the groups did not significantly differ on hypertension, hypercholesterolemia, or low blood pressure after adjusting for age and gender. The CADASIL group had worse cognitive performances, greater frequency of ischemic stroke, more psychiatric impairment, and decreased functional status compared to the SIVD group. Hypertension was correlated with worse functional status in the CADASIL group. Cognitive domains of memory, processing speed, and executive function were computed and were differentially related to functional status in the two groups. Functional status in the CADASIL group was significantly related to the memory domain, whereas it was significantly related to the executive domain in the SIVD group. The authors call for further study to elucidate the role of the memory in the functional status of CADASIL patients. The authors noted that O’Sullivan et al. [39] had previously shown a relationship between memory impairment and hippocampal atrophy in CADASIL, and that rather unlike SIVD, white matter lesions in CADASIL include the temporal lobes.
The foregoing review indicates that the cognitive features of CADASIL tend to coalesce around the cognitive domains of complex attention, processing speed, and executive functioning. These deficits likely arise because of a partial or, in the presence of lacunar infarction, complete disconnection. These same cognitive domains are the most consistently affected in sporadic subcortical ischemic vascular disease [40, 41] and disconnection is thought to underlie these deficits as well [42]. However, there is evidence that hippocampal atrophy is a key factor in the development of the dementia syndrome in patients with subcortical lacunar infarctions [43]. The role of the hippocampus in the development of dementia in CADASIL is uncertain. In a preliminary study, our group showed no difference in hippocampal volume between eight non-demented CADASIL patients and ten age-matched controls [44]. However, O’Sullivan and colleagues [39] demonstrated that hippocampal volume is an independent predictor of cognitive performance in CADASIL.
Magnetic resonance imaging features
Widespread diffuse WMH, indicating leukoencephalopathy, on T2-weighted MRI and T1-weighted hypo-densities are the imaging hallmarks of CADASIL (Figure 20.2). The WMH usually appear as punctate or nodular signals between the ages of 20 and 30 years [45], although individuals under 20 have not been extensively studied. Almost all gene-positive individuals will have evidence of WMH after age 30 [45]. Over time, the WMH become diffuse, symmetrically distributed, and may involve virtually all of the white matter. Cerebral microbleeds are also common in CADASIL, occurring in approximately one-third of individuals [18, 46, 47]. Intracerebral hemorrhage has been less frequently reported [48, 49, 50] and may be related to the number of cerebral microbleeds [49] and anticoagulant treatment [50].
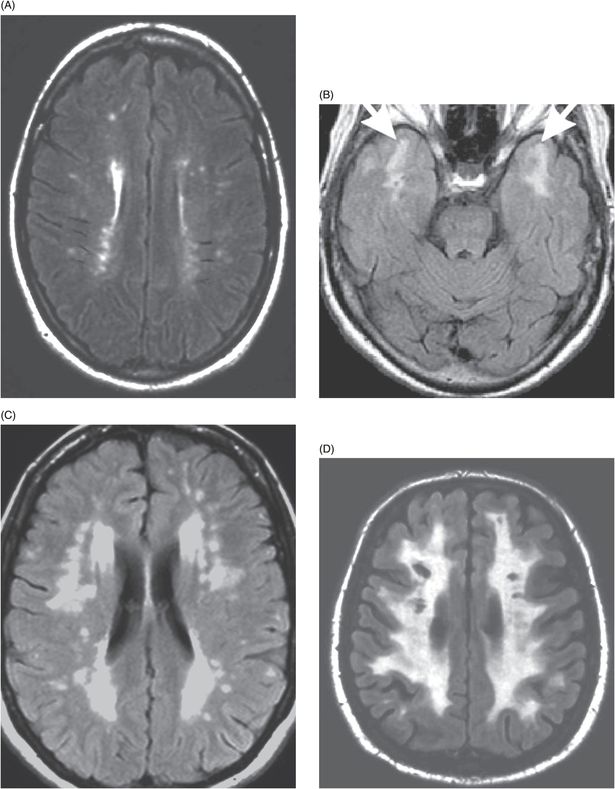
White matter hyperintensities on axial fluid-attenuated inversion recovery (FLAIR) magnetic resonance images in CADASIL. (A) A 25-year-old mutation carrier with no symptoms. (B) A 45-year-old with very early cognitive symptoms. Arrows show white matter hyperintensities in the anterior temporal lobes. (C) A 58-year-old with executive dysfunction and moderate areas of confluent white matter hyperintensities in the periventricular and deep white matter. (D) A 65-year old with dementia and extensive white matter hyperintensities with multiple lacunar infarctions.
The WMH occur most prominently in the centrum semiovale, frontal, temporal, and periventricular white matter [51]. They are also found in the internal and external capsules; corpus callosum; subcortical arcuate fibers; brainstem, particularly the pons and cerebellum; and in subcortical gray matter structures, including the caudate and lentiform nuclei and the thalamus [34, 51, 52, 53, 54]. Orbitofrontal and occipital white matter are relatively spared. Lacunar infarcts tend to have a similar distribution [34] but are somewhat less frequent in parietal, occipital, and infratentorial regions [55].
Microbleeds occur in CADASIL, particularly with increasing age [46, 47], occurring most commonly in the thalamus, basal ganglia, and brainstem [18, 46]: a distribution that contrasts with that of WMH and lacunes [18]. They have been shown to be associated with elevated systolic blood pressure, a history of hypertension, poor glucose control (indicated by glycosylated hemoglobin levels), and the volume of WMH and lacunar infarction [18].
The regional distribution of MRI lesions in CADASIL corresponds with the cerebral angioarchitecture [56]. The lesions are thought to arise from disruption of normal cerebral hemodynamics [56] resulting from the degradation of the smooth muscle layer, particularly in deep penetrating small arteries [51, 53]. Cerebrovascular reactivity (CVR) appears to play an important role. In one study, CADASIL patients demonstrated decreased CVR after administration of acetazolamide, as compared with controls, but not decreased total cerebral blood flow (TCBF). Follow-up imaging showed that the CADASIL patients had a larger increase in WMH but not lacunar infarcts. Furthermore, progression of lacunar infarcts and microbleeds was not associated with TCBF or CVR, possibly suggesting a different pathophysiology for the subcortical infarcts in CADASIL patients. The authors postulated that decreased CVR might predict CADASIL disease progression as demonstrated by increasing WMH [57].
Additionally, there is a positive correlation between age and both T1- and T2-weighted lesion load [53, 58, 59]. Opherk et al. [58] demonstrated a strong modifying effect of genetic factors on the MRI lesion volume in CADASIL. However, prior studies failed to find an association between the specific genetic abnormalities and MRI lesion load [59]. The discrepant results could reflect differences in the way that the genetic information was analyzed.
The MRI features of CADASIL overlap considerably, with the more common sporadic form of age-related SIVD related to hypertensive arteriosclerosis [60]. However, certain MRI features may help to differentiate the groups, particularly the presence of anterior temporal WMH, and extension of WMH into the cortical U-fibers [51]. Patients with CADASIL also tend to have greater WMH involvement in the superior frontal white matter [51] and in the external capsule and corpus callosum [54, 61], although these regions are less specific for CADASIL. Identification of WMH in the anterior temporal lobe and in the external capsule may also help to differentiate CADASIL from multiple sclerosis [62], a common misdiagnosis in CADASIL [63].
MRI studies in CADASIL have generally found associations between white matter lesion load and extent of functional and cognitive impairment. In a sample of 75 patients, Chabriat et al. [64] found that dementia only occurred in the presence of high-grade WMH. Using semi-automated techniques, Dichgans et al. [59] studied a group of 64 patients with CADASIL and found that both lacunar infarct and WMH volumes were significantly correlated with measures of functional disability and were inversely correlated with overall cognitive function based on the Mini Mental State Examination (MMSE). Liem et al. [26] and others [65, 66] demonstrated that lacunar lesion load is more predictive of cognitive dysfunction than either WMH or microbleeds. In contrast to these studies, a few reports in the literature with smaller groups of patients have failed to demonstrate an association between MRI lesions and extent of disability or cognitive impairment (e.g., Taillia et al. [33], Trojano et al. [67], Scheid et al. [68]).
Peters et al. [59] showed that the progression of WMH over time may be a less sensitive predictor of decline than loss of brain volume. They demonstrated significant loss of brain volume over a 2-year follow-up in a group of 76 patients with CADASIL. Age and hypertension were significant predictors of volume loss over the follow-up period but T2-weighted lesion load at baseline was not. Volume loss over the follow-up period was significantly associated with declines on clinical measures of stroke-related disability and on a structured dementia interview, but only a statistical trend was found for the association with the Mattis Dementia Rating Scale. Change in volume identified in T2-weighted images was not significantly correlated with change in any of these clinical measures These findings were corroborated by a two-center cohort trial that evaluated the effect of WMH, lacunar lesions, microbleeds, brain atrophy, and tissue microstructural changes on clinical impairment in 147 CADASIL patients. Brain atrophy was shown to be the strongest independent variable in predicting cognitive impairment [69].
In a cross-sectional study of 69 CADASIL patients, Jouvent et al. [70] found that measurements of cortical depth and surface area from 3D T1 MRI images strongly correlated with both cognitive and disability scales, independent of age and several potential cerebrovascular confounds (e.g., hypertension, body mass index, hypercholesterolemia). In a large longitudinal cohort study, Jouvent et al. [71] found further evidence of a relationship between changes in cortical morphology and worsening clinical status in CADASIL independent of global brain atrophy. Specifically, reduction of sulcal depth was independently associated with increased time to complete Trail Making Test parts A and B. Additionally, reduced cortical thickness was associated with increased disability, yet brain atrophy was only related to global cognitive worsening assessed by Mattis Dementia Rating Scale.
These findings suggest that, although subcortical infarction is the hallmark of CADASIL, intracortical lesions may play a role in the pathophysiology. The relationship between cognitive function and subcortical vascular changes may be complex, with contributions from cortical changes as well. Righart et al. [72] studied a sample of 98 CADASIL patients. Across the sample, per-decade declines in cortical thickness were nearly twice that reported for age-related atrophy among healthy individuals and the relationship between age and cortical thinning was greatest in prefrontal cortex. Cortical thickness declined significantly with increasing volumes of subcortical white matter lesions and was significantly correlated with processing speed in the left medial frontal cortex and the right occipitotemporal cortex. Bayesian network analysis showed that the left medial frontal cortex, but not the occipitotemporal cortex, had an intermediate position between lacunar lesions in the left anterior thalamic radiations and processing speed. Lesion load in the left anterior thalamic radiation was not directly significantly correlated with processing speed. This group of investigators had previously shown that a frontal–subcortical network, including the anterior thalamic radiations and the dorsolateral prefrontal cortex and cingulate circuits, underlie processing speed in CADASIL [73, 74]. Additionally, this group has shown that normalized white matter hyperintensity volume in the absence of lacunar infarcts in CADASIL patients correlates with performances on measures of processing speed, executive function, and attention but not with severe disability and that additional lacunar lesions are needed to produced significant cognitive impairment [65]. This latter finding aligns with that of Lee et al. (2011) [66] showing that the number of lacunes, but not normalized white matter hyperintensity volume, is the main factor predicting cognitive impairment in CADASIL.
Studies in CADASIL using MRI are limited by the sensitivity of conventional T2-weighted techniques to subtle white matter injury that nonetheless contributes to cognitive impairment [75]. Diffusion-tensor imaging (DTI) is more sensitive to disruption of white matter integrity than conventional MRI methods [76]. This technique provides indirect information about the structural integrity of white matter based on measurement of the magnitude and orientation of water diffusion in tissue [77]. Higher levels of diffusivity and lower levels of diffusion directional coherence (anisotropy) are interpreted as evidence of decreased white matter integrity. Diffusion-tensor imaging is an excellent technique for studying the full spectrum of white matter change in CADASIL.
Diffusion-tensor imaging studies in CADASIL have consistently found declines in white matter integrity compared with controls in both lesioned and normal-appearing white matter (NAWM) on T2-weighted imaging and in certain subcortical gray matter structures. These DTI changes correlate significantly with measures of disability and cognitive function. For example, Chabriat et al. (1999b) [64] found DTI changes in NAWM in patients with CADASIL compared with controls and these changes correlated significantly with a rating of disability but not with a global cognitive screening measure. O’Sullivan et al. (2004) [78] showed higher diffusivity in NAWM of non-demented patients with CADASIL compared with controls and these changes correlated with declines in executive function.
Changes in DTI have also been found in subcortical gray matter structures in CADASIL, including the thalamus, putamen, and globus pallidus [78, 79], and increased diffusivity in the thalamus correlates with executive dysfunction [78] and with performance on the MMSE [79]. O’Sullivan et al. (2005) [80] showed that executive function and verbal memory were associated with a distinct regional pattern of white matter changes in CADASIL: executive functions were correlated with decreased white matter integrity in distributed frontal white matter regions and in the cingulum bundle, whereas verbal memory ability was associated with DTI changes in the striatum only. Controlling for WMH volume did not appreciably alter the results, highlighting the importance in cognitive functioning of subtle changes in white matter integrity that are not visible on T2-weighted MRI. Diffusion-tensor imaging is also sensitive to the progression of white matter injury in CADASIL. Molko et al. (2002) [81] found that, compared with controls, patients with CADASIL had increased diffusivity in the entire brain image after 29 months and the change was associated with increased disability. A larger study also showed that increased diffusivity over a 2-year period was a significant predictor of worsening disability and cognitive decline in CADASIL but T2-weighted lesion volume was not [82].
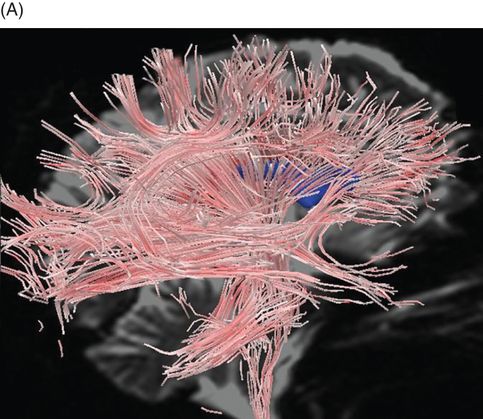
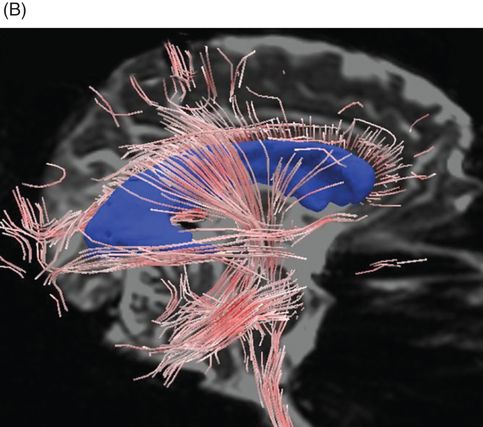
The foregoing studies involved analysis of images of scalar DTI parameters of diffusivity and anisotropy. These parameters consider the magnitude of diffusion in each image voxel. DTI tractography allows an alternative way to visualize DTI data that incorporates both the magnitude and direction of greatest water diffusion. Tractography visualizations provide a computer-generated representation of the three-dimensional topography of white matter architecture. We demonstrated the utility of quantitative DTI tractography in which measurements of the fiber models are used as markers of the structural integrity of specific fiber bundles (Figure 20.3) [83].
Diffusion-tensor imaging using streamtube models in normal elderly and CADASIL. (A) Whole-brain streamtube model (sagittal view) for a 72-year-old healthy volunteer. (B) Whole-brain streamtube model of a 60-year-old patient with CADASIL and mild dementia. Note the marked decrease in streamtube density in the patient with CADASIL. Streamtube models are superimposed on non-diffusion encoded T2-weighted magnetic resonance images; the lateral ventricles are portrayed in blue.
High-field in vivo MRA allows for evaluation of affected arterial structures and provides a vascular imaging complement to structural MRI methods. The ability of MRA to visualize the lenticulostriate arteries makes it ideal for studying CADASIL [84, 85]. Liem et al. (2010) [86] using 3D time-of-flight MRA showed no significant differences in luminal diameters in lenticulostriate arteries between CADASIL patients and controls, with no stenotic segments identified in CADASIL patients. In contrast to prior postmortem studies demonstrating mural thickening of leptomeningeal and lenticulostriate arteries, this imaging study found no association between lenticulostriate luminal diameter and lacunar infarct load in the basal ganglia or with basal ganglia hyperintensities. These results suggest that there may be an alternative pathophysiology for basal ganglia damage in CADASIL and that generalized narrowing of luminal diameters of lenticulostriate arteries may not play a significant role in the pathophysiology of CADASIL.
Pathology
CADASIL is associated with degeneration of small and medium-sized arterioles in subcortical white and gray matter. Figure 20.4 shows characteristic gross and microscopic brain changes in postmortem CADASIL. Granular material accumulates in the walls of the arterial smooth muscle layer and the presence of granular osmophilic material (GOM) adjacent to the basement membrane of the smooth muscle cells of cerebral arterioles on electron microscopy has become a hallmark pathological feature. As the disease progresses, GOM increases and cytoarchitectural changes include detachment of cells, with increasing space between the endothelium and the vascular smooth muscle cells (VSMCs) and disruption of the elastin and smooth muscle actin layers. Destruction of VSMCs may also cause decreased secretion of vascular endothelial growth factor (VEGF) and loss of vascular permeability [87, 88]. Severely damaged pericytes have also been found in CADASIL patients, leading to further disruption of cerebral microcirculation [89]. In arterioles, endothelial cells are swollen, leading to loss of tight junctions [87]. There is severe adventitial fibrosis from the site of the penetrating artery at the cortical surface to the distal end, transforming the vessel into an “earthen pipe” [90]. Over time, there is dilatation of the perivascular spaces, which appear as areas of signal hyperintensity on T2-weighted MRI. The subcortical white matter shows rarefaction, demyelination, and gliosis – more extensive but qualitatively similar to that seen in Binswanger’s disease. The progressive white matter injury is likely related to impairments in flow, reactivity, and autoregulation through the subcortical arterioles. However, we have demonstrated impairments in notch signaling in glial cells as well, suggesting that CADASIL is also a disease of the glia in subcortical white matter [91]. Large vessel infarctions are rarely seen. Lacunar infarction is common and occurs primarily in subcortical white and gray matter structures, though lacunar infarction also involves the brainstem. Autopsy examination of patients in their sixties who had late-stage disease reveals good preservation of the cortical ribbon on gross inspection, with widespread gliosis and demyelination of the subcortical white matter studded by numerous small holes in the white matter, which may represent areas where subcortical arterioles have dropped out.
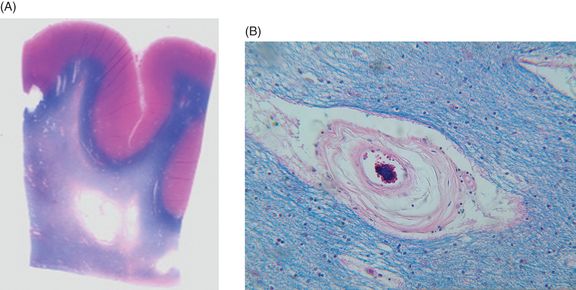
Gross and microscopic changes in postmortem CADASIL brain. (A) Luxol-fast blue, hematoxylin and eosin stain of the frontal lobe showing cavitary necrosis in the deep white matter with relative preservation of the subcortical U-fibers and normal thickness of the cortical ribbon. (B) Light microscopic hematoxylin and eosin stain of a subcortical arteriole demonstrating mural fibrosis and disintegration of the smooth muscle layer, dilatation of the perivascular space, and pallor of the surrounding white matter.
The actual cause of ischemic events and lacunar infarction in CADASIL is uncertain. CADASIL is not typically associated with hyalinosis, arteriosclerosis, luminal narrowing, atherothrombosis, or amyloid angiopathy, all of which are risk factors for typical age-related cerebrovascular white matter injury. Fragility and fragmentation of the vessel wall is the likely cause of microhemorrhage in later stages of the illness. As mentioned above, late-stage disease is associated with cerebral atrophy, and apoptotic changes have recently been reported in the cerebral cortex in postmortem tissue [18]. Fibrillar amyloid or tau pathology has only been reported in a single case but we have observed two additional cases of moderate Braak neurofibrillary pathology without amyloid plaques (unpublished data). Arteriolar changes are also observed in other organs such as the skin but symptoms related to vasculopathy in other organ systems, peripheral nerve and muscle are rarely reported [92].
Animal models of CADASIL
Postnatal Notch3 brain expression in mice is limited to vascular VSMCs within the walls of small to medium penetrating arteries, the same vessels predominantly damaged in CADASIL [92]. A mouse with an R142C Notch3 knock-in (corresponding to the common human CADASIL mutation R141C) did not show a CADASIL-like phenotype [93]. In 2003, however, a group of French researchers reported their creation of a transgenic mouse in which VSMCs expressed a full-length human Notch3 protein carrying the R90C change in low levels. By 10 months of age, the mice showed disruption of normal VSMC anchorage to the extracellular matrix of adjacent cells, VSMC cytoskeleton changes, and initial signs of VSMC degeneration. By contrast, Notch3 accumulation and GOM deposits did not appear until 14–16 months. (The mice did not develop brain parenchyma lesions or clinical symptoms.) The authors concluded that VSMC degeneration is not initiated by the buildup of Notch3 or GOM but perhaps by the disruption of VSMC anchorage [94]. They subsequently found that transgenic mice that had begun to exhibit cystoskeletal changes and disruption of adhesion of VSMCs to neighboring cells, but not accumulation of Notch3 and GOM, had impairment in pressure-induced contraction and flow-induced dilatation in isolated arteries; in other words, mechanotransduction (but not response to vasoactive agents) was impaired at an early stage in the disease, prior to Notch3 and GOM accumulation [95]. Mice at this early stage already showed impairment in reactivity to vasodilator stimuli and in cerebral blood flow autoregulation [96].
A transgenic mouse model was developed to analyze outcomes of mutations in the Notch3 binding domain ligand (EGFR10-11). Transgenic expression of C428S mutant NOTCH3 on a heterozygous knockout background inhibited downstream signaling by the normal Notch3 copy when compared to non-transgenic heterozygous knockout. These findings were then extrapolated to phenotypic significance in humans from a one large prospective cohort of 176 CADASIL patients. Ten patients from five distinct pedigrees, carrying a mutation in EGFR10 or 11, were identified. The mutation was associated with significantly higher MMSE scores and Mattis Dementia Rating Scale scores when compared with common mutations [38].
Genetic aspects of age-related SIVD
White matter hyperintensities as seen in age-related SIVD have been shown to be strongly heritable [97, 98, 99]. However, only limited work has been done on the specific genes that confer this risk apart from those known to impart general risk for hypertension, and even less work has been directed at the identification of genes that mediate the impact of vascular pathology on brain parenchyma [100]. Sierra et al. (2002) [101] found that, among 60 hypertensive patients (age 50–60 years) with white matter lesions, 64% had the DD genotype of the gene for angiotensin-converting enzyme compared with 22% of those without white matter lesions. In a large community-based sample of adults aged 44–75 years, Schmidt et al. (2000) [102] found that diastolic blood pressure and the LL genotype of the gene PON1, encoding paraoxinase, predicted the 3-year progression of white matter lesions. The association between apolipoprotein E genotype (APOE) and white matter lesions has been mixed, with some studies showing that allele type imparts a risk for white matter lesions [103, 104] and others showing no association [105, 106]. These mixed results could be partially explained by a finding from the population-based Rotterdam study suggesting that individuals with the APOE ε4 allele have increased risk for white matter lesions if they also have hypertension [107].
CADASIL genetics
Much more is known about the molecular genetics of the disease-causing mutations in CADASIL. Positional cloning and linkage analysis was used to locate the responsible gene on chromosome 19p13.1–13.2. Transmission of CADASIL Notch3 mutations is autosomal dominant with 100% penetrance. Notch 3 is a large gene comprising 33 exons that is ubiquitously found in human adult tissues, but its expression is restricted to vascular smooth muscle cells (Figure 20.5). The gene encodes a transmembrane protein of 2321 amino acid residues [12]. The function of Notch3 is to maintain cell–cell interaction or communication between vascular smooth muscle cells and arterial endothelial cells, thus maintaining arterial vessel homeostasis by promoting smooth muscle survival [108]. The Notch3 proteins are large, single-pass, transmembrane receptors. There is an extracellular domain that contains 34 tandem epidermal growth factor (EGF)-like repeats, three cysteine-rich Notch/Lin12 repeats, a single transmembrane domain and an intracellular domain. Each EGF repeat contains six conserved cysteine residues, which form three disulfide bonds, and almost all disease-causing mutations result from a missense mutation leading to a loss or gain of a cysteine residue in the EGF repeats encoded by the first 23 exons. The change in cysteine results in an odd number of cysteine residues and disruption of disulfide pairing. (Even the rare mutations that do not involve a cysteine directly may affect the location and function of neighboring cysteine residues [109].)
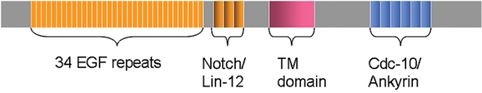
Structure of the Notch3 gene. Mutations occur in exons 3, 4, 11, and 18 in the epidermal growth factor (EGF) region in 80 % of carriers. Marcus et al. (2002) showed the majority of CADASIL patients (73 %) have mutations on exon 4. This produces an amino acid change associated with an increase or deletion of a cysteine. TM, transmembrane region.
Notch is processed in a similar manner to cleavage of the amyloid precursor protein, and inhibition of presenilin/γ-secretase also inhibits Notch. The Notch intracellular domain consists of several distinct units: a RAM domain is followed by a nuclear localization sequence, six ankyrin repeats (ANK), and a PEST domain, which is apparently involved in Notch receptor turnover [110, 111, 112]. Notch receptors bind DSL ligands (delta and serrate/jagged in Drosophila and vertebrates, Lag-2 in Caenorhabditis elegans). Like the Notch receptors, the ligands are single-pass transmembrane proteins with multiple EGFs in their extracellular domains [110]. In the Golgi apparatus, the full-length Notch3 protein is proteolytically cleaved by furine convertase into a 210 kDa extracellular fragment and a 97 kDa transmembrane and intracellular fragment (site 1 or S1 cleavage) [113]. The two fragments are non-covalently linked and transported to the plasma membrane as a heterodimeric receptor [114]. Upon ligand binding, S2 cleavage occurs at an extracellular site, allowing presenilin/γ-secretase-dependent S3 cleavage in the intramembrane domain and subsequent release of the soluble intracellular domain (NIc). This is then transferred to the nucleus, where it binds via the RAM domain and ANK repeats to the transcription factor RBP-Jk/CBF1 and converts it from a transcription repressor to a transcription activator [111, 112].
There is considerable phenotypic heterogeneity among family members with the same mutation, suggesting involvement of environmental and other genetic factors in phenotypic expression and significant relationships between genotype and phenotype have proven elusive. In a study of 127 CADASIL subjects from 65 families with 17 different mutations, no correlation was found between mutation and presence or age of onset of stroke, migraine, dementia, dependency or MRI lesion load, nor did particular families show specific phenotypes [20]. Figure 20.6 shows an example of phenotypic variability in terms of extent of WMH and clinical course between two members of the same family with the same Notch3 CADASIL mutation. Nevertheless, a few observations of specific genotype–phenotype correlations have been reported. The C117F mutation appears to be associated with a lower age at death and the C174Y mutation with a lower age at onset for stroke, immobilization, and death [22]. A Colombian family with the C455R mutation showed unusually early onset of stroke (median age 31 years; range 19–40) [115] and the R153C mutation has been identified as a risk factor for cerebral microbleeds in CADASIL [46]. A Japanese family with the S180C mutation had an unusual phenotype characterized by hallucinations and delusions and a decrease in the mean age at onset of stroke in each of three generations.
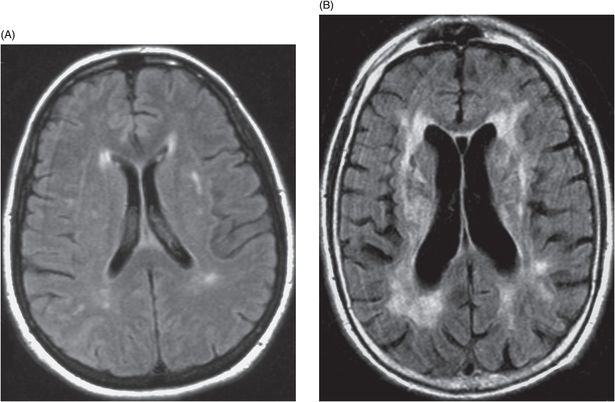
Phenotypic heterogeneity in a CADASIL family. (A) Axial fluid-attenuated inversion recovery (FLAIR) non-contrast magnetic resonance image (MRI) of a 55-year-old woman with mild subcortical white matter hyperintensities (leukoencephalopathy). She has had mild headaches but no significant cognitive, mood or motor symptoms. (B) Axial FLAIR non-contrast MRI of the brother of the patient in (A). Note the higher degree of leukoencephalopathy with multiple subcortical lacunar infarcts, as well as moderate cerebral atrophy and ventricular enlargement. This patient developed depression and recurrent ischemic episodes beginning at age 50, dementia at age 53, and died at age 55. The clinical course was complicated by alcohol abuse.
Molecular effects of mutations in CADASIL
Despite the thorough characterization of CADASIL-related Notch3 mutations, the pathophysiological mechanisms by which the mutations cause the vascular, histological and clinical effects of the disease are very poorly understood. The two main hypotheses for the pathogenesis of the disease are [1] accumulation of Notch3 protein products and GOM in the VSMCs, and [2] impaired signaling. Studies have variously supported and refuted both of these proposals.
The distribution pattern of Notch3 mutations suggests that the mutations exert a gain-of-function effect because they are located in areas of high sequence diversity among Notch orthologs [116]. These areas can tolerate significant diversity, presumably including those caused by CADASIL mutations, without a loss of function. Sites coding for signaling processes are, by comparison, usually highly conserved and suffer from loss-of-function mutations, so the findings of this study do not support a signaling deficit [116]. Furthermore, the 210 kDa extracellular fragment of the protein has been found to accumulate at the cytoplasmic membrane of cerebral VSMCs. Because production of Notch3 does not appear to be increased, the accumulation is probably caused by impaired clearance of the ectodomain from the cell surface, most likely as a result of improper oligomerization of the mutant protein fragment owing to a change in the tertiary structure or aggregation state of Notch3 [117]. Murine Notch3 cell lines with an R142C mutation (corresponding to the prevalent human mutation R141C) exhibited normal signaling but impaired processing, trafficking, and localization of Notch3 [118]. (However, later attempts to make a mouse model of CADASIL with this particular R142C mutation were not successful, so its validity as a model of human disease is unclear [93].) CADASIL-like mutations engineered into Notch3 in rats may cause a decrease in the ratio of receptor fragments to full-length protein but do not appear to affect signaling [119, 120]. Three-dimensional homology models of the first six EGF-like domains have suggested that some of the mutations would cause protein misfolding, perhaps leading also to homo- or heterodimeric intermolecular cross-linkage as a result of the uneven number of cysteine residues [121].
Two studies have found signaling impairment caused by mutations in the ligand-binding domain but not by mutations in EGF 2–5, the area of highest mutation density [37, 122]. However, in one of the studies, the mutant receptors, although targeted to the cell surface, showed decreased ratio of cleaved receptor fragments to full-length protein [37]. This result aligns with the findings of impaired trafficking and localization by Karlström et al. (2002) [118] discussed above. The finding that VSMC impairment precedes the onset of GOM and Notch3 accumulation in a mouse model of CADASIL seems to support a signaling hypothesis over an accumulation hypothesis [94, 95, 96].
Recent evidence implicates abnormal recruitment of extracellular matrix proteins by excess Notch3 in the pathogenesis of CADASIL. Monet-Lepretre et al. (2013) [123] analyzed cerebral and arterial tissue from CADASIL patients and mouse models of CADASIL. Using biochemical, nano-liquid chromatography-tandem mass spectrometry, and immunohistochemical analyses, the authors found that Notch3 aggregation in brain arterial vessels is a crucial event, leading to the abnormal recruitment of functionally important extracellular matrix proteins (tissue inhibitor of metalloproteinases 3 and vitronectin, which may result in multifactorial toxicity impairing extracellular matrix homeostasis in small vessels).
Related posts:
 The non-fluent/agrammatic variant of primary progressive aphasia
The cognitive neurology of corticobasal degeneration and progressive supranuclear palsy
The non-fluent/agrammatic variant of primary progressive aphasia
The cognitive neurology of corticobasal degeneration and progressive supranuclear palsy
 Amnestic mild cognitive impairment
Amnestic mild cognitive impairment
 The Lewy body dementias
The Lewy body dementias
 Semantic dementia (semantic variant primary progressive aphasia)
Semantic dementia (semantic variant primary progressive aphasia)
 Autoimmune antibody-associated encephalopathy and dementia syndromes
Autoimmune antibody-associated encephalopathy and dementia syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


