Fig. 1
Overview and simplified global classification of the main cognitive processes. See (Schmidt et al. 2007) for more detailed explanations
Most studies on the circadian modulation of cognitive function have focused on the impact of time of day on vigilance and basic attentional parameters (Schmidt et al. 2007). Historically, research revealed a temporal relationship between circadian variations in cognitive performance measures and daily fluctuations in physiological variables such as core body temperature. That is, when body temperature is high and endogenous melatonin is low, alertness and neurobehavioral performance tend to be higher (Kleitman et al. 1938). It was suggested that the circadian-related increase in body temperature would indirectly speed up cognitive processing by increasing metabolic activity in the brain (Kleitman et al. 1938). However, further research highlighted the role of other, external factors, on time-of-day effects in cognition . More specifically, peak and troughs in performance can be attributed to the type and difficulty of the task (e.g., differential working-memory load) (Folkard et al. 1983). While performance speed on simple repetitive and serial search tasks peaks with temperature levels in the evening (Colquhoun 1981; Monk 1982), speed performance on more complex cognitive tasks (e.g., logical reasoning tasks) peaks in the late morning (Folkard 1975), and performance in short-term memory retention peaks in the early to mid-morning (Laird 1925). Thus, Bonnet proposed that the optimal time of day for completing a cognitive test is largely dependent on the specific parameters of the task, such as its cognitive domain, duration and difficulty, the administration method, and the measured variable (Bonnet 2000). Alternative data, however, revealed that the selected paradigm (e.g., normal sleep/wake conditions vs. 40 h of enforced wakefulness during constant routine) also influences temporal performance (Cajochen et al. 1999). Moreover, compensatory mechanisms, such as motivational factors and expectancy due to experience, also play a role in the outcome (Schmidt et al. 2007).
To date, the picture that emerges is that time-of-day modulations affect performance on a range of cognitive tasks, and these performance fluctuations are additionally contingent upon inter-individual differences in circadian preference (i.e., chronotype). It seems that only highly practiced responses (e.g., constant performance tasks) (Valdez et al. 2005) are rather invariant across the day, with all other responses being vulnerable to the time-of-day effect during normal day–night conditions, as they require a certain degree of control over stimuli and responses. Above attentional processes, higher-order cognitive functions, such as working-memory load or executive control, appear to be particularly sensitive to time-of-day modulations (Mikulincer et al. 1989). However, given the current lack of research in this domain, and the varying choices of protocol and experimental control, it is impossible to conclude whether different tasks, involving a range of cognitive processes or differing in difficulty, exhibit genuine differences in time-of-day modulations (Schmidt et al. 2007).
3 Sleep Pressure Modulates Cognitive Performance
3.1 Sleep Deprivation Affects Attentional Processes
According to the “wake state instability” hypothesis (Doran et al. 2001), neurobehavioral performance becomes increasingly variable under the influence of elevated sleep pressure due to inadvertent microsleep episodes, with brief moments of low arousal that make it difficult to sustain attention. This unstable state, which fluctuates from second to second, is characterized by increased lapses of attention, increased numbers of errors in response, but also increased compensatory efforts resulting in normal reaction times for a short period of time. Over the last two decades, the instrument that has emerged as the dominant assay of vigilant attention in paradigms of sleep loss is the PVT (Dinges et al. 1985). This task has been widely used in human studies to detect the sustained attention (or “vigilance”) deficits associated with different types of sleep loss, including chronic sleep restriction (Belenky et al. 2003; Van Dongen et al. 2003) and sleep deprivation (Doran et al. 2001; Rétey et al. 2006). Importantly, the task is highly sensitive to sleep loss, independent of aptitude, lacks learning effects, and its reliability and validity have been amply demonstrated (Lim and Dinges 2008). The PVT is a test of simple reaction time to a cue that occurs at random inter-stimulus intervals. During the task (standard duration of 10 min) subjects are instructed to attend to a small, rectangular area on a dark screen. They are then required to respond as quickly as possible whenever they perceive the appearance of a bright millisecond counter inside this rectangular area. Stopping the counter allows subjects to view their reaction time, which serves as feedback for that particular trial. Button presses, when the counter is not displayed on the screen, are counted as false starts, which subjects are instructed to avoid. Four dominant findings have emerged from the use of the PVT in sleep research protocols. First, sleep deprivation results in an overall slowing of responses. Second, sleep deprivation increases the propensity of individuals to lapse for lengthy periods (>500 ms), as well as make false starts. Third, sleep deprivation enhances the time-on-task effect, the phenomenon whereby performance worsens across the course of a cognitive task owing to fatigue and reduced motivation. Finally, PVT results during extended periods of wakefulness reveal the presence of interacting circadian and homeostatic sleep-regulatory processes (Lim and Dinges 2008).
3.2 Sleep Deprivation Affects Higher-Order Cognitive Processes
Sleep deprivation has been shown to have significant adverse effects on a range of higher-order cognitive processes, including memory encoding, consolidation, and retrieval (Walker 2008), behavioral inhibition (Drummond et al. 2006; Harrison et al. 2007), judgment (Killgore et al. 2007a), planning (Horne 1988; Killgore et al. 2009), and divergent thinking capacities (Horne 1988). All such processes are believed to draw heavily upon resources in the prefrontal cortex (Killgore et al. 2011). Moreover, recent research from rodent experiments has highlighted that sleep deprivation is associated with reduced neural activity within brain regions involved in memory (frontal cortex and hippocampus), emotion (amygdala), and regulation of the sleep–wake cycle (anterior hypothalamus and supraoptic nucleus) (Hagewoud et al. 2011; Pierard et al. 2007; Vecsey et al. 2009). Disruption of any of these distinct facets of cognition by sleep deprivation may contribute to noteworthy errors in decision making (reviewed by Killgore 2010). Interestingly, however, deficits in executive functions have not been observed universally, particularly during shorter durations of sleep deprivation, such as one night (Pace-Schott et al. 2009). This suggests that the brain’s executive function systems may temporarily compensate for brief sleep loss by utilizing additional cognitive resources via activation of alternative brain regions (Drummond et al. 2000, 2005b).
More recently, research has focused on clarifying the ways in which sleep deprivation may influence well-characterized, higher cognitive processes, such as mental heuristics and emotional biases that affect risk assessment and decision making (Killgore et al. 2012). For example, McKenna and colleagues (2007) revealed that when the possible outcomes from a gambling task were framed in terms of potential gains, sleep deprivation prompted subjects to take more risks compared to when they were well rested. Yet, when the same task was presented in terms of potential losses, lack of sleep led them to take fewer risks than usual. Such findings indicate that sleep deprivation may lead to greater reliance upon preexisting cognitive biases. Moreover, functional neuro-imaging studies have highlighted that sleep-deprived individuals show differences within brain-reward circuitry during risky decision making, and this may bias them toward expectations of gains while reducing their focus on losses (Venkatraman et al. 2007).
Another way that prolonged wakefulness affects decision making is by reducing the weight that a person places on new information when making choices (Dickinson and Drummond 2008). Sleep deprived individuals may tend to rely more upon automatic, as opposed to effortful, forms of cognitive processing (Killgore et al. 2012). Emotional biasing is a form of automatic processing that may influence decision making. Indeed, Damasio (1994) proposed that emotional reactions act as a cognitive streamlining function that quickly and efficiently narrows an individual’s choice of options. These emotional “gut reactions” prime a person to make choices based on how rewarding or unpleasant they found a previous similar experience. In an experimental setting, this emotion-guided decision making can be investigated using the Iowa Gambling Task (IGT) (Bechara et al. 1994). During the computerized program, participants are presented with four decks of cards placed face down. Next, players are required to select 100 cards from these four available packs. On card selection, they are immediately informed as to whether the card they selected results in a monetary gain or a monetary loss. Unbeknownst to the subject, however, two of the decks are “good” decks and lead to small but consistent net gains; while the other two decks are “bad” decks, and comprise large short-term gains but consistent long-term losses. With regard to the results, healthy individuals usually learn from the trial-by-trial feedback and adjust their playing strategy to avoid the risky bad decks in favor of the modest, but consistently advantageous, good decks (Bechara 2004). However, patients with damage to the ventromedial prefrontal cortex (vmPFC) fail to make this adjustment (Bechara 2004). Such findings are in line with evidence that damage to the vmPFC leads to shortsightedness for the future (Bechara et al. 1994), as well as neuroimaging data that indicates that this brain region plays a key role in the decision making process of the IGT (Li et al. 2010).
Importantly, the vmPFC seems to be particularly affected by sleep deprivation. Metabolic activity in this brain region is drastically reduced after a single night of sleep loss (Thomas et al. 2000), whereas increased activation of this area is correlated to a subject’s degree of responsiveness to rewards during sleep-deprived decision making (Venkatraman et al. 2011). Accordingly, in a series of studies performed by Killgore and colleagues (2006, 2007b), the IGT was used to assess the effects of sleep deprivation on emotionally-guided decision making. As predicted, well-rested subjects rapidly learned the contingencies of the task. However, following 49 (Killgore et al. 2006) and 75 h (Killgore et al. 2007b) of prolonged wakefulness, the same participants showed a significant decline in decision making performance. Specifically, they became progressively more risk-taking and short-sighted in decision making, tending to prefer risky short-term gains at the expense of incurring long-term losses. Overall, such findings indicate that prolonged sleep loss is associated with making choices that begin to favor short-term over long-term outcomes—a pattern paralleling that often observed among patients with lesions to the vmPFC (Bechara 2004). Since the vmPFC is important in several key cognitive-affective processes (Damasio 1994), alterations in vmPFC functioning, or its associated neuro-circuitry following sleep loss, may indeed underlie some of the subtle changes in decision making observed in the two Killgore et al. studies. The findings are also in accordance with evidence suggesting that sleep deprivation leads to difficulty incorporating new information into ongoing decision making processes, implying an overall decline in cognitive flexibility, in favor of greater reliance on automatic cognitive processes (Dickinson and Drummond 2008).
Taken together, these behavioral findings suggest that distinct higher-order cognitive processes are impaired by sleep deprivation. The sensitivity of the prefrontal cortex to the effects of sleep loss may also be reflected in distinct neurophysiological changes associated with sleep deprivation. For example, regional cerebral blood flow in this region correlates with electroencephalogram (EEG) slow-wave activity (SWA; power density in the 0.75–4.5 Hz range) in non-rapid-eye-movement (NREM) sleep (Dang-Vu et al. 2010), which represents the primary physiological marker of sleep homeostasis (Achermann and Borbély 2011). Moreover, not only the increase in SWA in NREM sleep, but also the rise in EEG theta (~5–9 Hz range) activity after prolonged wakefulness (Cajochen et al. 1995) is larger over anterior than over posterior cortical areas (Finelli et al. 2000).
4 Cerebral Underpinnings of Circadian and Homeostatic Influences on Performance
Accumulating evidence demonstrates that circadian and homeostatic sleep–wake regulatory processes interact in a fine-tuned manner to modulate cognitive performance (Schmidt et al. 2012). Neural connections from the SCN indirectly reach target areas implicated in sleep homeostasis, including ventro-lateral-preoptic area, tuberomammillary nucleus, lateral hypothalamus, thalamus, and brainstem nuclei via its connections to the dorsal medial hypothalamus (Mistlberger 2005). Simultaneously, diffuse monoaminergic activating systems are under circadian control and adjoin with many thalamo-cortical areas, which suggests that the interaction with sleep homeostasis takes place at many different levels (Dijk and Archer 2009).
Research conducted by Aston-Jones and colleagues indicated that the noradrenergic locus coeruleus plays an important role in the circadian regulation of arousal (2001, 2005). Activity in the locus coeruleus, combined with its widespread thalamic and cortical connections, may modulate a variety of central nervous system functions, including alertness and vigilance, and also higher-order cognitive processes (Cajochen et al. 2010). Moreover, a recent study incorporating behavioral assessments, EEG, and functional magnetic resonance imaging (fMRI) in morning and evening chronotypes indicated that homeostatic sleep pressure exerts an influence on attention-related cerebral activity in key structures crucially involved in generating the circadian wake-promoting signal, including the locus coeruleus. Specifically, maintenance of optimal attentional performance in the evening after a normal waking day was associated with higher activity in evening chronotypes than in morning chronotypes in locus coeruleus and anterior hypothalamus, including the SCN (Schmidt et al. 2009). Furthermore, activity in the anterior hypothalamus decreased with increasing homeostatic sleep pressure, as indexed by EEG SWA in the first NREM sleep episode. These data suggest that circadian and homeostatic interactions contribute to the neural activity that underlies diurnal variations in human behavior. Interestingly, the differential activation pattern was observed only for optimal performance on the PVT (i.e., the fastest 10th percentile of reaction times) (Schmidt et al. 2012), which reflects the phasic ability to recruit the attentional network above normal levels (Drummond et al. 2005a).
The mechanisms by which circadian oscillations in the SCN, as well as circuits controlling for states of wakefulness and sleep, interact at the cerebral level in order to regulate arousal and cognitive behavior, are yet to be clarified (Cajochen et al. 2010). Conceptually, endogenous “sleep substances” may accumulate during wakefulness and modify activity in key areas regulating cortical arousal, including brainstem, hypothalamic nuclei and basal forebrain. During sleep, the “sleep substances” would dissipate. Although the biochemical “substrate” of sleep homeostasis remains poorly understood, adenosine, nitric oxide, prostaglandin D 2, tumor necrosis factor alpha, interleukin-1, growth-hormone-releasing hormone, and brain-derived neurotrophic factor are considered to be important candidate mediators of the consequences of prolonged wakefulness (Krueger et al. 2008).
5 A Role for Adenosine in Homeostatic Sleep–Wake Regulation
Compelling and converging evidence in animals and humans has accumulated over the past two decades to support a role for adenosine and adenosine receptors in sleep–wake regulation (see A et al. 2008; Landolt 2008; Porkka-Heiskanen and Kalinchuk 2011 for reviews). Animal studies suggest that the extracellular adenosine concentration in the brain may increase during prolonged wakefulness and decline during (recovery) sleep (Porkka-Heiskanen et al. 2000).
5.1 Adenosine Formation, Transport, and Metabolism
The formation of adenosine in the brain changes in an activity-dependent manner and different mechanisms contribute to the appearance of adenosine in extracellular space. Increased energy demand during wakefulness leads to the break-down of energy-rich adenine nucleosides such as adenosine-tri-phosphate (ATP). Adenosine is formed in neurons by 5′-nucleotidase and transported through plasma and intracellular membranes by specialized transporters, including sodium-driven concentrative, and equilibrative nucleoside transporters (Fig. 2). The concentrative transporters use energy to move adenosine into the cell, whereas the equilibrative transporters transport adenosine according to the extracellular/intracellular concentration gradient. Elevated intracellular adenosine concentrations following increased utilization of ATP in conditions of high energy demand lead to release of adenosine. In addition, extracellular adenosine is also formed by ecto-nucleotidases through hydrolysis of ATP. Release of ATP from synaptic vesicles occurs along with several other neurotransmitters, including the major excitatory neurotransmitter glutamate (Haydon and Carmignoto 2006). Finally, ATP and glutamate are also released from astrocytes by a recently established process referred to as gliotransmission. Molecular genetic manipulations in mice strongly suggest that glial cells provide a significant source of extracellular adenosine in the brain (Haydon and Carmignoto 2006). Furthermore, astrocyte-derived ATP may activate purinergic (e.g., P2X7) receptors and affect sleep independently from adenosine (Krueger et al. 2008, 2010).
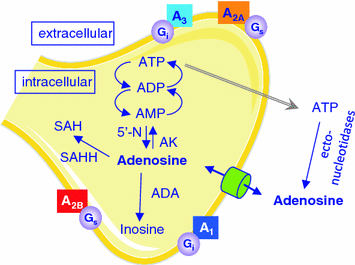
Fig. 2
Schematic representation of adenosine formation, metabolism, and transport. Neurons, astrocytes and microglia cells can release adenosine and adenosine-tri-phosphate (ATP; gray arrow). All cell types express adenosine receptors , adenosine transporters (cylinder), and ecto-nucleotidases that convert ATP into adenosine. A 1, A 2A, A 2B, A 3: adenosine receptors coupled to corresponding G-proteins; ADP: adenosine-di-phosphate; AMP: adenosine-mono-phosphate; SAH: S-adenosyl-homo-cysteine; 5′-N: 5′-nucleotidase; AK: adenosine kinase; ADA: adenosine deaminase ; SAHH: S-adenosyl-homocysteine hydrolase
Clearance of extracellular adenosine mostly occurs through the non-concentrative nucleoside transporters (Fredholm et al. 2005). The main intracellular metabolic pathways of adenosine are the formation of adenosine-monophosphate by adenosine kinase, and the irreversible breakdown to inosine by adenosine deaminase (ADA). Ecto-ADA also catalyzes the extracellular deamination of adenosine. Mainly due to the high activity of adenosine kinase, baseline levels of extracellular adenosine usually remain low. The action of ADA, which appears to be more abundantly expressed in astrocytes than in neurons (Fredholm et al. 2005), may be particularly important when elevated concentrations of adenosine have to be cleared, such as after sleep deprivation. Both, molecular genetic manipulations of adenosine kinase in mice (Palchykova et al. 2010), as well as genetically reduced ADA enzymatic activity in humans (Rétey et al. 2005), increase deep slow wave sleep and EEG SWA in NREM sleep. These findings provide strong additional support to the idea that adenosine importantly contributes to the homeostatic control of sleep.
5.2 Adenosine Affects Neuronal Systems Regulating Wakefulness and Sleep
Adenosine attenuates the activity of wakefulness/vigilance-promoting neurons in brainstem (e.g., locus coeruleus), basal forebrain (BF), and hypothalamus (e.g., tuberomammillary nucleus) and may contribute to cortical disfacilitation, a form of inhibition due to reduced activating input from ascending cholinergic and monoaminergic pathways. As suggested by intracellular recordings in non-anaesthetized cats the long-lasting hyperpolarizing potentials in NREM sleep, which provide the cellular substrate of EEG SWA, may represent periods of disfacilitation (Steriade et al. 2001; Timofeev et al. 2001). Moreover, adenosine activates neurons of the hypothalamic ventro-lateral-preoptic area by reducing inhibitory γ-amino-butyric-acid (GABA)-ergic inputs. These neurons fire significantly faster after sleep deprivation than they do during normal sleep, indicating that their activity is modulated by homeostatic mechanisms representing sleep need (Sherin et al. 1996).
One current hypothesis based upon biochemical, pharmacological, electrophysiological, and behavioral studies postulates that elevated adenosine in the BF plays a distinct role in mediating the sleep deprivation-induced increase in sleepiness and homeostatic sleep drive (Basheer et al. 2004; Porkka-Heiskanen and Kalinchuk 2011; Strecker et al. 2000). It may be important to note, however, that Blanco-Centurion and colleagues highlighted that the actions of adenosine are not restricted to the BF region (2006). This research team used a lesion and pharmacological approach to reveal that adenosine accumulation in the BF is not necessary for sleep induction, and also that BF cholinergic neurons are not essential for sleep drive. Thus, the available data rather suggest that extracellular adenosine provides a global feedback signal on a neuronal network, including subcortical and cortical structures (Franks 2008), that regulates important functional aspects of wakefulness and sleep.
6 Adenosine A1 and A2A Receptors Mediate Effects of Adenosine in Sleep–Wake Regulation
The cellular effects of adenosine are mediated via four subtypes of G-protein coupled adenosine receptors : A 1, A 2A, A 2B, and A 3 receptors. In vitro studies indicate that physiological concentrations of endogenous adenosine can activate A 1, A 2A, as well as A 3 receptors. Nevertheless, it is widely accepted that the high-affinity A 1 and A 2A receptors are primarily involved in mediating the effects of adenosine on sleep and vigilance, at least in humans (Sebastiao and Ribeiro 2009).
6.1 Adenosine A1 Receptors and the Effects of Prolonged Wakefulness
The stimulation of A 1 receptors opens several types of K+-channels, inhibits adenylate cyclase through activation of Gi proteins and inactivates transient voltage-dependent Ca2+-channels. The A 1 receptor is ubiquitously, but not homogenously, expressed in the central nervous system (Bauer and Ishiwata 2009). In vivo PET with the selective A 1 receptor antagonist, 18F-CPFPX, revealed highest receptor occupancy in striatum and thalamus, as well as temporo-parietal and occipital cortex (Fig. 3a). Pre- and post-synaptic activation of A 1 receptors inhibits excitatory neurotransmission. This receptor subtype, therefore, has long been assumed to play an important role in sleep–wake regulation. Pharmacologic and genetic studies in rats and mice, as well as molecular imaging in humans, partly support this notion. For example, inducible knock-out of neuronal A 1 receptors in mice reduces SWA (3.0–4.5 Hz range) in NREM sleep under baseline conditions, and attenuates the homeostatically regulated rise in SWA after sleep restriction (Bjorness et al. 2009). Moreover, prolonged wakefulness appears to up-regulate A 1 receptor binding in subcortical and cortical brain structures in animals and humans (Elmenhorst et al. 2007, 2009). Taken together, these data indicate a role for adenosine A1 receptors in mediating distinct consequences of sleep deprivation.
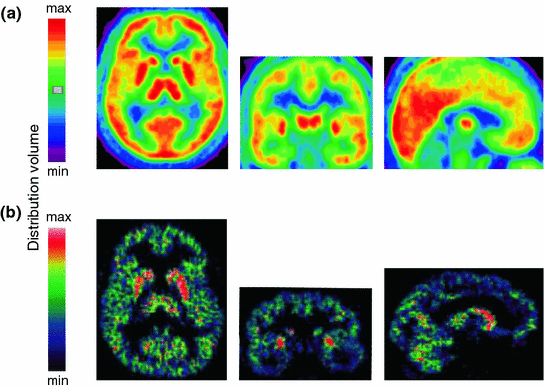
Fig. 3
Distribution of adenosine A 1 and A 2A receptors in the human brain. a Color-coded distribution volumes of the selective A 1 receptor antagonist, 18C-CPFPX (mean values of 10 healthy young men). From left to right: axial, coronal, and sagittal planes (coordinates according to the Montreal Neurological Institute brain atlas: z = −4, y = −12, x = 0) (Unpublished data). b Color-coded distribution volumes of the selective A 2A receptor antagonist, 11C-KW6002 (istradefylline), in a healthy male volunteer. From left to right: axial, coronal, and sagittal sections. Figure modified from (Brooks et al. 2008)
6.2 Adenosine A2A Receptors and the Effects of Prolonged Wakefulness
The stimulation of A 2A receptors increases adenylate cyclase activity through activation of G s (or G olf in striatum) proteins, induces the formation of inositol phosphates, and activates protein kinase A. Compared to the A 1 receptor, this adenosine receptor subtype is less widely distributed in the brain (Bauer and Ishiwata 2009). The highest expression in the human central nervous system is found in basal ganglia (particularly in putamen and caudate nucleus) (Fig. 3b). Recent studies in rodents, including experiments in knock-out mice, suggest that also A 2A receptors contribute to the effects of adenosine on sleep. Local administration of the selective A 2A receptor agonist, CGS21680, to the subarachnoid space adjacent to BF and lateral preoptic area increases c–fos expression in the ventro-lateral-preoptic area and promotes NREM sleep (Scammell et al. 2001). Direct activation of sleep-promoting neurons of the ventro-lateral-preoptic region upon stimulation of A 2A receptors could underlie this effect (Gallopin et al. 2005). Interestingly, preliminary data suggested that mice with A 2A receptor loss-of-function have reduced sleep and an attenuated sleep rebound after sleep deprivation (Hayaishi et al. 2004), indicating that A 2A receptors are part of the neural network that regulates sleep homeostasis in mammals. These findings are supported by recent data in humans, suggesting that genetic variants of the A 2A receptor gene (ADORA2A ) modulate the sleep deprivation-induced increase in EEG SWA in NREM sleep (Bodenmann et al. 2012; Landolt 2012).
In conclusion, both adenosine A 1 and A 2A receptor subtypes probably mediate functional effects of adenosine after sleep deprivation, whereas distinct effects may be site- and receptor-dependent.
7 Adenosine and Sleep-Associated Cognitive Functions
7.1 Sleep Deprivation, Adenosine, and Vigilant Attention
While a wealth of evidence supports the concept that modulation of cerebral adenosine contributes to the regulation of wakefulness and sleep, it was not until more recently that research revealed how this manipulation could also alter neurobehavioral performance (Christie et al. 2008). This is poignant given that the BF in particular has been implicated not only in adenosinergic mechanisms of sleep regulation but also in the control of sustained attention (Baxter and Chiba 1999). Thus, the fact that decrements in sustained attention tend to occur concomitantly with feelings of sleepiness , is consistent with studies indicating that the same mechanisms implicated in the control of the homeostatic sleep drive, are also involved in the regulation of attention (Zaborszky et al. 1997). Moreover, neurons within the BF project to components of the cortical sustained attention network, whose activation is linked with optimal human performance on the PVT (Drummond et al. 2005a). More recently, a rat version of the PVT was developed that enabled invasive investigations of the role of adenosine and the BF in the control of behavioral state and sustained attention (Christie et al. 2008). Christie and colleagues (2008) utilized this task to assess the effects of elevated cerebral adenosine on vigilant performance. The study revealed that rats receiving infusions of adenosine in the BF immediately prior to performing the PVT showed prolonged response latencies and more performance lapses. The effect was blocked by the co-administration of the A 1 receptor antagonist, 8-cyclopentyl-theophylline, demonstrating that the performance decrements were indeed due to elevated adenosine in the BF and apparently mediated by A 1 receptors, as opposed to other, unrelated factors (Christie et al. 2008). Furthermore, the adenosine-induced impairments in sustained attention were similar to those seen in rats undergoing sleep deprivation (Cordova et al. 2006). These findings are consistent with the hypothesis that sleep loss induces an accumulation of adenosine in the BF, which leads to increased sleepiness and reduced vigilance.
Local cerebral administration of adenosine is not possible in humans, to study its role in reduced vigilant attention during sleep deprivation. Nevertheless, relevant information has been obtained from studies on a naturally occurring genetic variation of the gene encoding the adenosine metabolising enzyme ADA. A G-to-A single nucleotide polymorphism at nucleotide 22 of the gene encoding ADA underlies an Asp-to-Asn amino-acid substitution at codon 8 of the ADA protein. Compared to G/G homozygotes, carriers of the variant allele show reduced ADA activity in vitro (Battistuzzi et al. 1981; Riksen et al. 2008), and presumably elevated tissue adenosine levels in vivo (Hirschhorn et al. 1994). This functional polymorphism not only modulates the duration and intensity of slow wave sleep (Bachmann et al. 2012; Mazzotti et al. 2012; Rétey et al. 2005), but also human attentional performance in rested and sleep-deprived states. More specifically, carriers of the G/A genotype (n = 29) performed worse on the d2 focused attention task than G/G homozygotes (n = 191) (Bachmann et al. 2012). The difference was also present between two prospectively matched subgroups of G/A (n = 11) and G/G (n = 11) genotypes. Moreover, sustained attention (Fig. 4a) and vigor were reduced, whereas waking EEG alpha activity (8.5–12 Hz), sleepiness, fatigue, and a-amylase activity in saliva were enhanced in A-allele carriers when compared to G/G homozygotes. These convergent data demonstrate that genetic reduction of ADA activity in healthy humans not only modulates the quality of sleep, but also the quality of wakefulness, including neurobehavioral performance.
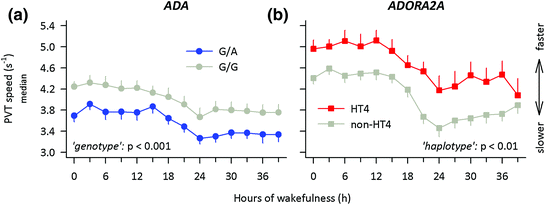
Fig. 4
Functional variants of genes contributing to adenosine metabolism (adenosine deaminase, ADA) and signal transmission (adenosine A 2A receptor, ADORA2A ) contribute to inter-individual differences in psychomotor vigilance during prolonged wakefulness. Starting 30 min after wake-up from the baseline night, a 10-min psychomotor vigilance task (PVT) was administered at 3-h intervals during 40 h prolonged wakefulness. Ticks on the x-axis are rounded to the nearest hour. The time courses of median speed (1/reaction times) are illustrated; error bars indicate + or − 1 SEM. Data were analyzed with 2-way ANOVA models with the factors ‘genotype’ (G/A, G/G) or ‘haplotype’ (HT4, non-HT4) and ‘session’ (14 assessments during prolonged waking). a Blue circles: G/A genotype (n = 11). Grey circles: G/G genotype (n = 11). The G/A genotype of ADA performs worse than the G/G genotype throughout prolonged waking (‘genotype’: F 1, 25 = 15.4, p < 0.001; ‘session’: F 13, 239 = 38.6, p < 0.001; ‘genotype’ × ‘session’ interaction: F 13, 146 = 0.3, p > 0.9). Data were re-plotted from (Bachmann et al. 2012). b Red squares: Carriers of HT4 haplotype alleles (n = 6). Gray squares: Carriers of HT4 haplotype alleles (n = 17). See (Bodenmann et al. 2012) for details of genetic analyses. Individuals with haplotype HT4 performed faster than non-HT4 allele carriers throughout sleep deprivation (‘haplotype’: F 1, 21 = 9.3, p = 0.006; ‘session’: F 13, 273 = 16.3, p < 0.001; ‘haplotype’ × ‘session’ interaction: F 13, 273 = 0.9, p > 0.5). Data were reanalyzed from (Bodenmann and Landolt 2010)
7.2 Sleep Deprivation, Adenosine , and Higher-Order Cognitive Functions
As discussed previously, Bjorness and colleagues (2009) revealed that conditional knock-out of the A 1 receptor elicits selective attenuation of the SWA rebound following restricted sleep. This research team also investigated the effects of this genetic manipulation on working memory. It demonstrated that animals lacking the A 1 receptor not only showed a reduced rebound SWA response, but they also failed to maintain normal cognitive function, although this function was normal when sleep was not restricted. Since the attenuation of SWA is associated with compromised working memory performance, this indicates a functional role for adenosine A 1 receptor-dependent SWA homeostasis in maintaining this cognitive ability when sleep is restricted (Bjorness et al. 2009). Here, it is worth noting that the loss of the A 1 receptors in the conditional gene deletion used in this study is exclusively neuronal. Nevertheless, the source of the adenosine includes both neuronal and non-neuronal, or glia, cells (Pascual et al. 2005; Studer et al. 2006). Halassa and colleagues (2009) inhibited the release of adenosine from glia cells in transgenic mice expressing a dominant-negative (dn) SNARE domain in astrocytes, in order to investigate if astrocytes play a role in sleep–wake regulation. They found that this genetic restriction of gliotransmission attenuated the build-up of sleep pressure, and prevented memory deficits associated with sleep loss. The data suggest an important role for astrocyte-derived adenosine in modulating cognitive consequences of sleep deprivation. Furthermore, the research team also conducted pharmacological studies and concluded that astrocytes modulate the accumulation of sleep pressure and its cognitive consequences through a pathway involving adenosine A 1 receptors (Halassa et al. 2009).
8 The Adenosine Receptor Antagonist Caffeine and Sleep-Loss-Associated Cognitive Impairments
Caffeine is the most widely consumed stimulant in the world. In the μM plasma concentrations reached after moderate consumption (Landolt et al. 1995; Carrier et al. 2007), caffeine acts as a non-selective, competitive antagonist at A 1 and A 2A receptors (Fredholm et al. 1999). Novel PET imaging findings suggest that intake of 4–5 cups of coffee (corresponding to ~450 mg caffeine) in a 70-kg volunteer can displace endogenous adenosine from 50 % of cerebral A 1 receptors (Elmenhorst et al. 2012). By contrast, other effects of caffeine observed in vitro, such as inhibition of phosphodiesterase, blockade of GABAA receptors and Ca2+ release, require more than 100 times higher doses than adenosine receptor antagonism and are toxic in humans (Fredholm 1995).
8.1 Caffeine Counteracts Sleep Deprivation-Induced Impaired Vigilant Attention by Interfering with Sleep Homeostasis
Various studies have examined the effects of caffeine on sustained attention in humans, via performance on the PVT. The psychostimulant has been consistently shown to reverse sleepiness and PVT impairments in sleepy humans (Landolt et al. 2004; Balkin et al. 2004; Kamimori et al. 2005; Rétey et al. 2006; Van Dongen et al. 2001; Wyatt et al. 2004; Landolt et al. 2012). Given that physiological doses of caffeine antagonize adenosine receptors , such findings are consistent with those of the aforementioned rodent study, which revealed decrements in vigilant performance following adenosine administration (Christie et al. 2008).
Wyatt and colleagues (2004) reflected that many studies investigating the neurobehavioral benefits of caffeine during sleep loss confounded the two major processes regulating sleep and wakefulness: the circadian phase and the duration of prior wakefulness (i.e., homeostatic sleep pressure). Specifically, previous research had not clarified if caffeine’s ability to counteract performance deficits related to sleep deprivation was related to its interaction with circadian or homeostatic signals modulating sleep propensity and performance. The authors stress the importance of accounting for variance explained by sleep homeostatic and circadian modulation when interpreting data from protocols in which tests are given in only a single administration, such as typically occurs in traditional clinical and cognitive neuroscience research. As a result, the research team conducted a study to assess the effects of repeated low-dose caffeine administration during a 29-day forced desynchrony paradigm. The period of the sleep–wake cycle was scheduled to be 42.85 h (28.57-h wake episodes and 14.28-h sleep episodes), and thus far removed from the circadian range. This protocol allowed for separate quantification of the circadian, sleep homeostatic, and caffeine contributions to performance deficits and improvements. Moreover, the 42.85-h cycle simulated the extended wakefulness commonly encountered by medical and military personal, or anyone skipping a night of sleep (Wyatt et al. 2004).
During the study, caffeine was administered during wakefulness at a rate of 0.3 mg per kg per hour. The dosage schedule was designed to increase caffeine blood plasma concentrations in parallel to the rate of increase in sleep homeostatic drive during wakefulness, and also in line with the potential accumulation of adenosine (Porkka-Heiskanen et al. 2000). Polysomnographic recordings were used to monitor each scheduled sleep episode, as well as the majority of each wake episode, in order to detect incidences of slow eye movements and unintentional sleep onsets. During wake periods, mood, and subjective sleepiness were assessed at 30-min intervals using visual analog scales and the Karolinska Sleepiness Scale. Cognitive performance was tested every 2 h.
Related posts:
 Memory Consolidation in Healthy Aging and Mild Cognitive Impairment
Restricted or Disrupted Sleep as a Causal Factor in the Development of Depression
Memory Consolidation in Healthy Aging and Mild Cognitive Impairment
Restricted or Disrupted Sleep as a Causal Factor in the Development of Depression
 and Electrophysiological Correlates of Sleep and Sleep Homeostasis
and Electrophysiological Correlates of Sleep and Sleep Homeostasis
 Treatment of Sleep Disorders and Its Relationship with Neuroplasticity
Treatment of Sleep Disorders and Its Relationship with Neuroplasticity
 and Synaptic Homeostasis
and Synaptic Homeostasis
 Studies of Sleep and Memory in Humans
Studies of Sleep and Memory in Humans
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


