Figure 16.1. An episode of sustained monomorphic ventricular tachycardia.
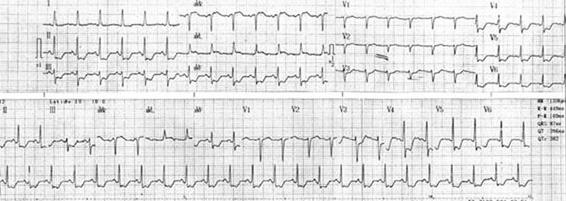
Figure 16.2. 36-year-old patient with traumatic brain injury and inferolateral ST-T depression without angiographically significant coronary lesions.
16.3 Paroxysmal Essential Hyper- and Hypopothassemia (Periodic Paralysis)
These kinds of paralysis are characterized by weakness episodes associated with increased potassium, though presenting normal K levels. Frequently, the attacks are induced by exercise and exposure to cold weather. Ventricular extrasystoles, ventricular tachycardia and ventricular bidirectional tachycardia are often observed.
Weakness episodes and arrhythmias usually respond to measures initiated to normalize plasma potassium and mexiletine levels.
Paroxysmal essential hypopothassemia manifests as periodic paralysis attacks, with low potassium levels. Andersen’s syndrome refers to periodic paralysis sensitive to potassium, associated with low implantation of the ears, clinodactyly, prolonged QT interval and ventricular arrhythmias. Weakness episodes are intense and prolonged, with attacks induced by exposure to cold weather and post-exercise rest. Carbohydrates can also cause episodes.
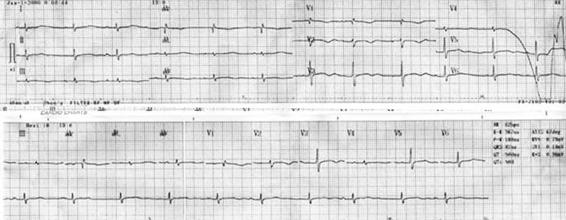
Figure 16.3. Sinus rhythm with prolonged QT interval.
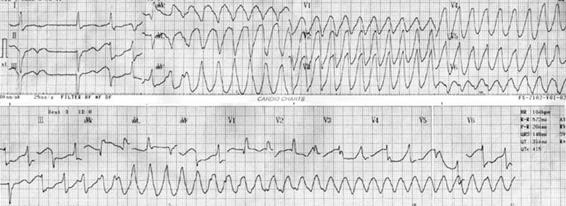
Figure 16.4. Torsades de pointes. Patient with severe hypokalemia, 1.6 mEq/l paralysis. Strong ventricular arrhythmias, QT prolongation and ventricular extrasystoles are frequent.
Correction of potassium levels usually improves the weakness, although arrhythmias tend to persist. Beta blocker therapy produces an improvement in non-sustained ventricular tachycardia. Anti-arrhythmia 1A-type medications should not be given because they exacerbate weakness and associated arrhythmias with prolongation of QT interval.
16.4 Dystrophy
Duchenne muscular dystrophy can present:
- Conduction disorders with an increase in conduction speed and short P-R (<120 msec.) or prolonged infranodular conduction in some patients.
- Rhythm disorders with sinus tachycardia secondary to alterations in the autonomic system, or due to fibrosis and fat infiltration of the sinus node, which develops a field for intranodal re-entrance; fibrillation and auricular flutter; ventricular extrasystole; ventricular tachycardia with late potential development. Heart death in 25% of patients, equally distributed between sudden death and progression to cardiac insufficiency.
Arrhythmia treatment with anti-arrhythmia drugs produces major respiratory insufficiency, besides a major risk of respiratory insufficiency associated with implantation of a heart defibrillator, without improvement in prognosis.
16.5 Myotonic Muscular Dystrophy
Cardiac alterations are mainly arrhythmias, with frequent ECG anomalies including AV blocks, branch blocks, and unspecified delays in intraventricular conduction. It manifests as pathological changes, with unknown mechanisms leading to conduction system degeneration (fibrosis and fat infiltration) of the sinus node, the AV node, the His and Purkinje fibres, and the auricular and ventricular fibres. It may progress to symptomatic myocardiopathy. Reduced conduction with positive late potentials is observed.
Conduction system disorders can progress to complete AV block, requiring a definitive pacemaker. Prophylactic implantation of a pacemaker must be evaluated. 2008 ACC/AHA/HRS guidelines suggest considering, in conduction disorders of myotonic muscular dystrophy patients, implantation of definitive pacemaker in patients with second or third degree symptomatic or asymptomatic AV block.
Auricular tachycardia, auricular flutter and auricular fibrillation are frequent arrhythmias (4%), often well-tolerated by controlled ventricular frequency due to concomitant conduction disorder.
Sudden death occurs in one third of patients and is believed to be caused by secondary asystole, although sudden deaths have been seen in pacemaker-bearing patients. Ventricular arrhythmias have been associated with branch-branch tachycardia.
16.6 Steinert Myotonic Dystrophy
This autosomal dominant disease is caused by a gene on the long arm of chromosome 19. It is clinically characterized by muscular weakness and myotony. Two forms are distinguished: a moderate, slowly progressive form with stationary periods, in which muscular weakness is limited to the forearms and dorsal foot flexors, scarce myotony without CNS disorders, while cataracts and cardiac conduction disorders are frequent; and the classical form characterized by more rapid progression and concomitant florid myotony.
Muscular weakness has a characteristic distribution (facial, mandibular, cervical and peripheral flexor); there is usually palpebral ptosis and weakness in the masseter and orbicular muscles. Unlike scapular-humeral dystrophy, the scapular waist is affected late in the course of the disease; and unlike other myopathies, distal muscular weakness in the extremities is worse. Myotony tends to appear between the age of 5 and 25 years and it is characterized by involuntarily maintained muscle contraction when hit by a reflex hammer, for example, to the thenar musculature or when inviting the patient to contract a muscle. These patients tend to have low IQ, apathy, aggressiveness, hypersomnia, possibly due to an increase in gliosis, and decreased blood flow as detected by proton spectroscopy studies.
Cardiac alterations are caused by progressive fibrosis in the conduction ways, shown as arrhythmias (first degree AV-block, sinus bradycardia and branch blocks); there is a major risk of sudden death in the early phase of the illness.
Arrhythmias often prompt patients to seek medical attention. It is important to perform periodical evaluation with cardiac echo-Doppler and ECG.
16.7 Emery-Dreyfuss Muscular Dystrophy
This familiar disorder presents with mild weakness in skeletal musculature and frequent and lethal arrhythmogenic disorders. A recessive hereditary disease (less frequently dominant) related to chromosome X, it generates an alteration in the membrane protein emerin. This protein is located in the cardiac desmosomes and is associated with conduction disorders.
The dystrophy presents a triad of precocious contractures of the posterior cervical muscles, elbow muscles and the Achilles tendon, with weakness and slowly progressive muscular atrophy and cardiac alterations.
It is associated with dilated myocardiopathy and a high prevalence of conduction disorders. Conduction disorders, with AV blocks, auricular flutter, auricular fibrillation, sinus arrest, and union-rhythm bradycardia, start to appear in the third and fourth decades of life. Cardiac stimulation becomes necessary by the age of 40-50 years.
Sudden death has been described in association with ventricular tachycardia and auricular fibrillation.
Follow-up and surveillance of ECG disorders by means of Holter monitoring, ECG and echocardiography should be done. With progression of the disease, stimulation with a permanent pacemaker and/or an implantable automatic cardiac defibrillator is recommended.
16.8 Muscular Dystrophy of the Extremities and the Waist
This group of hereditary recessive (type 1) and dominant (type 2) disorders, including subgroups 1B, 2C, is associated with cardiac alterations such as dilated myocardiopathy. Other disorders manifest with the evolution towards progressive AV block, requiring pacemaker implantation. Episodes of sudden death have been described, even with the use of a permanent pacemaker, which suggests ventricular arrhythmias.
Follow-up with ECG, Holter monitoring and echocardiography is recommended, with special attention to at-risk subgroups, and eventual pacemaker implantation.
16.9 Friederich’s Ataxia
This cerebrospinal degenerative recessive autonomic hereditary disease is characterized by ataxia of the extremities and torso, dysarthria, loss of deep tendon reflexes, sensorial anomalies, skeletal deformities and cardiac alterations. The genetic disorder is associated with chromosome 9, responsible for production of the protein frataxin.
The severity of the neurological symptoms and cardiac hypertrophy are related to alterations in this protein, which is a mitochondrial protein key to iron homeostasis and cell respiration.
Concentric ventricular hypertrophy is most frequently associated with Friederich’s ataxia, although it has been observed with asymmetric septal hypertrophy and/or dilated myocardiopathy in some cases. Concentric ventricular hypertrophy signs are the most frequent ECG findings, with diffuse T-wave inversion. Auricular arrhythmias, as auricular flutter or auricular fibrillation, are generally associated with progression of the disease.
Sudden death has been described, although its mechanism is not yet well known. Conduction disorders have not been described.
16.9.1 Treatment
Evolution to a severe dilated myocardiopathy requires treatment of cardiac insufficiency without any specific treatment.
16.10 Kearns-Sayre Syndrome
This encephalopathy results from anomalies in mitochondrial function. Histopathological studies of the skeleton and heart muscles show an abnormal proliferation of mitochondria.
Conduction alterations constitute the most typical cardiac abnormality, dilated myocardiopathy being less frequent. It presents with fibrosis and fat infiltration of the conduction system.
Diagnosis is based on the triad ophthalmoplegy, pigment retinopathy and cardiac block, with prolonged HV interval. Preexcitation leading to supraventricular paroxysmal tachycardia has been described. When there is a conduction disorder, prophylaxis with a definitive pacemaker is recommended.
16.11 Leber’s Hereditary Optical Neuropathy
Another mitochondrial encephalopathy, characterized by a painless loss of bilateral vision, this pathology is associated with a short PR interval and preexcitation syndromes, with the development of supraventricular arrhythmias.
Preexcitation with paroxysmal tachycardia symptoms indicates treatment of the anomalous conduction.
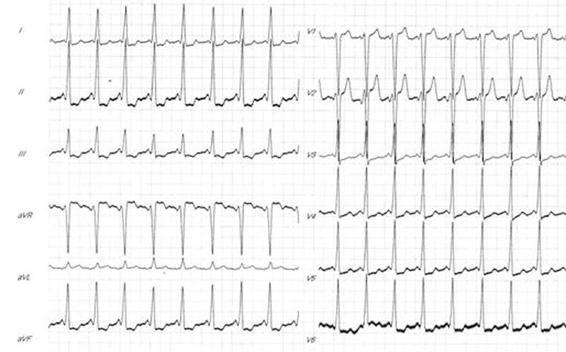
Figure 16.5. Preexcitation syndrome. Sinus rhythm, short PR and delta wave.
16.12 Guillain-Barré Syndrome
This is an acute demyelinating inflammatory neuropathy, with malfunction of peripheral nerves, cranial pairs and the autonomous nervous system. Arrhythmias such as bradycardia, asystole, inappropriate sinus tachycardia, auricular fibrillation, tachycardia and ventricular fibrillation are related to the autonomous disorder. It may also be associated with hypertension and diaphoresis. These arrhythmias lead to an increase in mortality.
Intravenous administration of immune globulin or plasmapheresis improves prognosis. Monitoring is important. In patients with rhythm disorders, implantation of a temporary or permanent pacemaker is recommended. Infusion of atropine or isoproterenol before tracheal aspiration manoeuvres is recommended.
16.13 Myasthenia Gravis
This chronic autoimmune neuromuscular disease is caused by the production of antibodies directed against the nicotine receptor of acetylcholine. Producing weakness in voluntarily-controlled muscles, this weakness increases with activity and decreases with rest. The main symptom is fluctuating weakness, which usually starts with the ocular and facial muscles. It is generally associated with thymus gland hyperplasia.
Myocarditis is frequently associated, although arrhythmias such as auricular fibrillation, AV block, asystole, and sudden death have also been observed
Immunosuppressive medication and anti-cholinesterase are part of the typical treatment. These can diminish heart rate and hypotension. Administration of uinidine and propranolol can exacerbate weakness.
16.14 Epilepsy
The estimated annual incidence of epilepsy and sudden death varies between 1/370 and 1/1000 epileptic patients. The incidence is higher in the 20-to-40-year age group and higher still in very symptomatic epileptic patients. Unexpected sudden death is 3.8 to 9.3 times more frequent in epileptic patients than in the general population: 100% of cases experience generalized tonic-clonic seizures alone or combined with another type of crisis, most complex partial crises having been deduced from what witnesses or doctors said or what the electroencephalogram showed.
Some 60% victims present with a structural cerebral lesion responsible for the attack. This percentage is much lower in non-selected epileptic patients. The types of lesion found were old frontal contusion, old penetrating lesion, chronic subdural hematoma, cortical arteriovenous malformation, tumour or old stroke.
Electrical events during an epileptic attack lead to centres involved in the heart’s neurovegetative function, causing cardiac arrest or fatal arrhythmia. It seems that sympathetic, more than parasympathetic, activity reaches the heart, causing a fatal cardiac response associated with the crisis, but it is unclear why this occurs in certain patients. It could be because of a subtle myocardial lesion difficult to demonstrate following habitual autopsy techniques. Some authors have reportedly found small fibrotic areas in the terminal arteries, focal lesions of the myocarditis type and focal endocardiac fibrosis in conduction bundles. These can have their origin, as shown by experimental studies, by intense repeated sympathetic stimulation during the crisis.
According to Lathers et al., electrophysiological evidence of cerebral discharge due to arrhythmogenic potential was induced by generalized crises in anesthetized and mechanically-ventilated cats. Recording two separate branches of the sympathetic cardiac nerve and the vagus nerve, they proved that activity outbreaks occurred in the sympathetic cardiac nerves synchronized with ictal and interictal points of the EEG. With determined doses of the substance used to induce the crises (penthylentetrazol, [PTZ]), the degree of synchronization, variable between both branches of the sympathetic cardiac nerve, could be lost. Therefore, it was suggested that the different branches of the sympathetic cardiac nerve innervate different areas of the ventricular myocardium. The imbalance in the activities of the sympathetic and parasympathetic nervous system branches is thought to provoke a non-homogeneity in heart depolarization and repolarization which allows for the appearance of irritability places, re-entrance circuits and ventricular ectopias. Furthermore, the authors suggested that this mechanism can be activated in a normal crisis in the presence of subtherapeutic levels of anti-epilepsy medication, resulting in malignant arrhythmias and sudden death.
The most important data have been obtained, nevertheless, by stimulation of the hypothalamus, although there are changes in arterial pressure and respiration. However, it seems difficult to believe this to be the highest cerebral level of cardiac representation: there should be cortical mechanisms involved.
As to the type of arrhythmia observed, there were auricular fibrillation, multifocal premature ventricular contraction, ventricular tachycardia, sinus bradycardia, asystole periods and torsades de pointes. Interestingly, we have a patient with a right frontal glioma repeatedly suffering from supraventricular paroxysmal tachycardia which disappeared only after tumour removal. An alternative explanation to this is that the high levels of catecholamines have their role in the production of a neurogenic pulmonary edema observed after the appearance of different brain lesions. Hypoxia conditions could generate acidosis and, consequently, arrhythmias.
Systematic studies of cardiac rhythm alterations during a crisis indicate the appearance of:
Related posts:
 Neuroscience Critical Care: Two Experts’ Point of View
Neurocritical Care of Patients With Arteriovenous Malformations
Neuroscience Critical Care: Two Experts’ Point of View
Neurocritical Care of Patients With Arteriovenous Malformations
 Pathophysiology of Intracranial Hypertension
Concepts and Management of Brain Death and Management of Potential Organ Donation
Pathophysiology of Intracranial Hypertension
Concepts and Management of Brain Death and Management of Potential Organ Donation
 Extracranial Atherosclerotic Carotid Artery Disease
Extracranial Atherosclerotic Carotid Artery Disease
 Surgical Treatment of Spinal Cord Injury
Surgical Treatment of Spinal Cord Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


