during wakefulness) may be an important variable in the development of central apneas during sleep (12). In patients with markedly diminished or absent chemosensitivity, some form of sleep-disordered breathing (SDB), frequently central apnea, would be expected. If carbon dioxide sensitivity is very low or absent, there would be little ventilation stimulus during sleep and central apneas with sustained hypoventilation would be predictable. Patients with disorders such as central alveolar hypoventilation (“Ondine’s curse”) and the obesity-hypoventilation (Pickwickian) syndrome would fall into this category (13,14 and 15). Similarly, recent reports have suggested a high prevalence of CSA among patients chronically taking narcotic medications (which can inhibit chemosensitivity) (16).
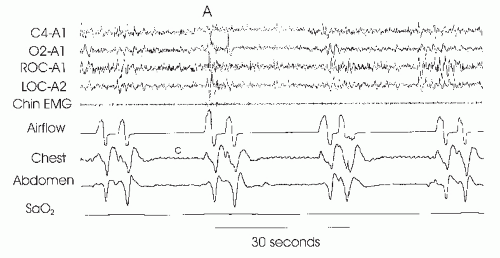 FIGURE 23-1 Idiopathic central apnea. The figure illustrates three apneas (cessation of airflow) without respiratory effort detectable in the chest and abdominal belts. Termination of the first apnea is associated with arousal (A). The small deflections in the chest tracing during apnea (c) do not reflect respiratory effort but are secondary to cardiac pulsations. |
TABLE 23-1 CLASSIFICATION OF CSA SYNDROMES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
levels during wakefulness and a high hypercapnic ventilatory response (17). The unusually steep hypercapnic responsiveness can lead to respiratory instability with a fluctuating pattern of ventilation in the wake—sleep transition. This instability is believed to be secondary to intermittent overventilation (yielding hypocapnia) alternating with underventilation (yielding hypercapnia). The cyclic nature of ventilation probably results from the marked fluctuations in ventilation that occur with modest changes in PaCO2. Thus, elevated chemosensitivity contributes to cessations in airflow by producing ventilatory overshoots during which PaCO2 falls below the apnea threshold.
TABLE 23-2 PATHOPHYSIOLOGICAL MECHANISMS OF CSA | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
the stimulation of J receptors in the lung, which also increases ventilatory drive. Thus, a prolonged circulation time probably must be combined with an increased ventilatory drive to yield (CSR) (39). Patients with CHF and CSR tend to maintain their waking eupneic PCO2 levels during sleep, thus keeping the PaCO2 close to the apnea threshold. Therefore, rather minor overshoots in ventilation could dramatically reduce ventilatory drive (40,41). These mechanisms (circulation time, CO2 responsiveness, CO2 set point) explain much of the variability in CSR occurrence in heart failure patients.
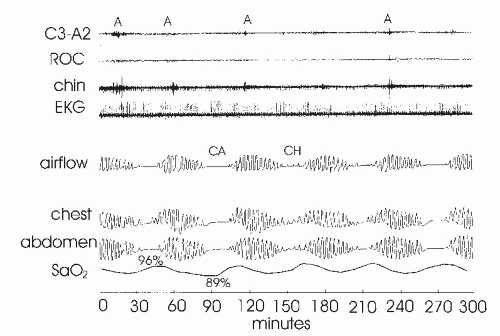 FIGURE 23-2 Cheyne-Stokes respiration. The figure illustrates a waxing and waning pattern of airflow and respiratory effort over the course of this 5-minute recording. Central apnea (CA) or central hypopnea (CH) occur at the nadirs in respiratory effort. Of note, the duration of each cycle is roughly 1 minute. The periods of ventilation between events are much longer than in idiopathic central apnea (Fig. 23-1). In addition, note the fall in oxygen saturation that occurs following each apnea. The time duration from the end of apnea to the nadir in arterial oxygen saturation (Sao2)-desaturation is prolonged and reflects circulatory delay. Although difficult to see at this resolution, there is frequently arousal (A) from sleep at the peak of the hyperpnea with associated tachycardia. In idiopathic central apnea, arousal usually occurs at apnea termination (Fig. 23-1). |
respiration to maintain ventilation. When these muscles develop the atonia characteristic of REM sleep, the diaphragm alone may be unable to maintain adequate ventilation. On the other hand, there does exist the rare patient with central defects in ventilatory control who experiences improvements in respiration during REM sleep, as compared with NREM sleep. This phenomenon is thought to result from the nonmetabolic influences on ventilation that occur in REM sleep (e.g., dream content) but not NREM sleep. However, this REM-related improvement is relatively uncommon.
apneas, spontaneous arousals) may increase the number of central apneas. Treatment-emergent central apneas (also referred to as complex apnea (48,49 and 50)) occur during the titration of continuous positive airway pressure (CPAP) in obstructive sleep apnea (OSA) patients (Fig. 23-4). Although this has been studied minimally and the explanation remains obscure, CPAP does lower upper airway resistance, which can lead to falls in PCO2. If the PCO2 falls below the apnea threshold, central apneas will ensue (51). Increased lung volume on CPAP may also activate stretch receptors that can inhibit ventilation. Clinical experience and emerging data suggest that this form of central apnea generally resolves with ongoing therapy. A similar phenomenon has been observed in OSA patients following tracheostomy, wherein some individuals will experience central apneas that eventually resolve (52). Patients with idiopathic CSA have no other identifiable disorder (i.e., diagnosis of exclusion). However, this syndrome is relatively rare (53).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


