Phases of development for a surrogate marker of progression in Parkinson’s disease (PD). Biomarker discovery leads to identification of species that are most often first studied in cross-sectional cohorts to provide information on disease association and also disease characteristics. This needs to be followed by characterization of biomarker performance in longitudinal cohorts. To validate a surrogate marker of PD progression, the biomarker will finally need to be tested in longitudinal clinical trials in which it is compared with a meaningful clinical outcome to determine that it will potentially substitute for that outcome in future clinical trials.
Targeted markers
Dopamine-based markers
Although measuring dopamine directly in fluids has not proved useful as a PD biomarker, studies of CSF have identified altered levels of related compounds. Particular attention has been paid to CSF homovanillic acid (HVA) levels, as HVA is the breakdown product of central dopamine (Figure 24.2). The ratio of HVA:xanthine is significantly different between the CSF of patients with PD and that of controls, and changed with time in one longitudinal study, suggesting its potential as a progression marker [13, 14]. In contrast, other potential dopamine-related markers are lacking longitudinal data that would provide information on changes with disease status. These include platelet monoamine oxidase B activity, responsible for dopamine breakdown, which has been found to be increased in the plasma of patients with PD [15]; reduced dopamine content, tyrosine hydroxylase immunoreactivity, and expression and density of dopamine receptors in peripheral blood lymphocytes [16]; and dopamine D2 and D3 receptor transcripts [17].
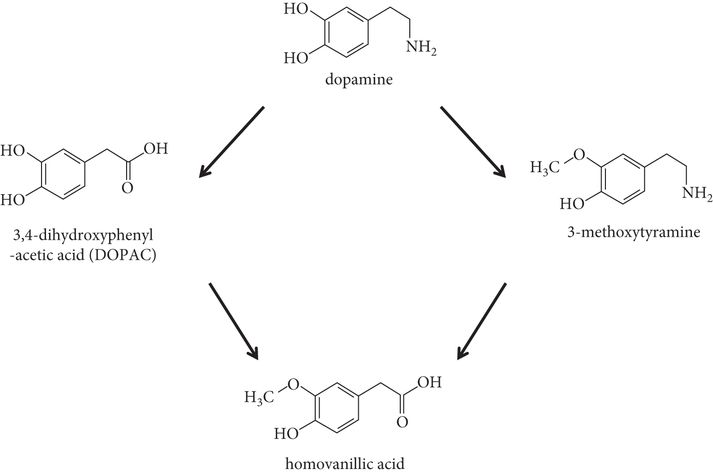
Dopamine metabolism and the relationship of homovanillic acid, a potential Parkinson’s disease biomarker, to dopamine.
Disease-related protein biomarkers
Advances in our understanding of PD etiopathogenesis have now identified a number of proteins that are key in cellular pathways perturbed in the disease. The first gene determined to lead to PD, α-synuclein (SNCA), allowed identification of a protein in which oxidative damaged and misfolding is now thought to be the major molecular factor underlying PD pathogenesis in the majority of cases. Multiple other genes have now been identified in which mutations either lead to or increase the risk of PD. Their transcripts and protein products are now under intensive investigation as potential biomarkers.
α-Synuclein
α-Synuclein is a major component of Lewy bodies and Lewy neurites, which represent the pathological hallmarks of PD. Although the focus has been on Lewy bodies present in surviving dopaminergic neurons of the substantia nigra, these structures are widespread within the central nervous system (CNS), and in peripheral autonomic nerves [18, 19], in salivary glands [20] and within skin tissue [21]. At least some such changes may occur in the “premotor” phase of PD, making α-synuclein a future candidate for monitoring prior to diagnosis on the basis of motor symptoms (“phenoconversion”). Although primarily intracellular, α-synuclein is detectable in plasma [22–24] and CSF [25].
Unfortunately, measuring α-synuclein in blood samples may be more difficult than anticipated, due to different forms that may be measured, as well as technical confounders. α-Synuclein total level [26], oligomeric form [22] and phosphorylated α-synuclein [24] have all been found to be elevated in blood in PD, but there are differences among studies. Others have found a trend for decreased α-synuclein [27] and Li et al. [28] found a greater decrement in α-synuclein in early-onset PD versus late-onset PD when measured by western blotting. Over 95% of α-synuclein in blood samples is thought to be located in red blood cells, with 1% or less in plasma, and therefore even a small variation in hemolysis would have a large confounding effect on results.
Measuring α-synuclein in CSF has been more robust. Decreased α-synuclein levels in CSF of individuals with PD versus controls have now been documented in a number of independent studies [25, 29–36]. Although the majority of these studies involved more advanced PD patients, similar results have been found in early PD compared with control subjects [35, 36], demonstrating that such a difference is not due to unanticipated medication treatment effects. How specific this finding will be to PD is uncertain. In one study, a lower level of α-synuclein was measured in CSF from Alzheimer’s disease (AD) versus controls, leading the investigators to postulate that it is a marker of loss of synapses in general [29], as opposed to a PD-specific process. A more recent study found decreased CSF α-synuclein not only in PD but also in dementia with Lewy bodies (DLB) and multiple-system atrophy (MSA) [34]. It will likely be important to examine different species of α-synuclein to obtain the full picture and appreciate its potential usefulness as a biomarker. Another important issue to consider is whether a combination of protein measurements, incorporating α-synuclein, might constitute a superior biomarker. In one study, a panel of seven proteins comprising α-synuclein, DJ-1, total tau, phosphorylated tau, fractalkine, Flt3 ligand, and β-amyloid 42 aa isoform differed in CSF samples from PD when compared with normal controls, AD and MSA [37].
Whether α-synuclein will ultimately turn out to be helpful in tracking PD progression remains to be seen. However, in the PPMI study [8], decreased α-synuclein was associated with increased motor severity in PD [35]. Moreover, a recent study has suggested that lower α-synuclein levels may be associated with a longitudinal cognitive decline, based on post-hoc analysis of CSF α-synuclein levels in samples stored from the DATATOP study of selegiline and α-tocopherol in PD [38].
Immunotherapy targeting α-synuclein has recently been investigated as a potential PD treatment. Active immunization with recombinant human α-synuclein reduces α-synuclein accumulation in neuronal cell bodies and synapses in a mouse model of PD, and was also demonstrated to reduce neurodegeneration in that model [39]. A vaccine, PD01A (AFFITOPE®; AFFiRiS), is now in an extension study following a phase 1 clinical trial. Passive immunization with an anti-α-synuclein monoclonal antibody has also been shown to reduce motor deficits and reduce α-synuclein expression on synapses and axons in an animal model [40]. It is thought that these effects occur through inhibiting the early stages of α-synuclein aggregation, thus preventing the development of oligomeric deposits [41]. Given these promising preclinical results and current translational initiatives, it is tempting to speculate that biomarkers centered on α-synuclein may be helpful in monitoring the response to intervention, and particularly in demonstrating target engagement, as has been described for AD markers [42–45].
DJ-1
Mutations in the DJ-1 gene are a rare cause of autosomal-recessive forms of PD. It is detected in blood and CSF. Similarly to α-synuclein, only a small fraction of DJ-1 is present in plasma compared with blood cells and platelets, making variable hemolysis or residual platelets in sample preparation a potential confounder [27]. No significant difference between PD versus control plasma levels of DJ-1 was detected by Maita et al. [46]. However, another study demonstrated a significantly higher concentration of plasma DJ-1 in PD compared with control subjects, and plasma levels of DJ-1 correlated with PD disease severity [47]. A recent large study of DJ-1 in CSF found decreased DJ-1 levels associated with PD, with 90% sensitivity and 70% specificity for the diagnosis of PD [30]. This study also measured α-synuclein and found that α-synuclein and DJ-1 levels in CSF were correlated. However, there are some differences in findings between groups, and a previous study actually suggested that DJ-1 levels were increased in CSF in PD, particularly in early stages [48]. Moreover, DJ-1 may not be a marker of disease severity [27, 37]. The picture is likely to be more complicated, since DJ-1 exists in multiple isoforms. Lin et al. [49] found that of seven isoforms detected in blood, those with posttranslational 4-hydroxy-2-nonenal modifications were altered in late PD. Additionally, a form of DJ-1 oxidized at cysteine-106 is elevated in red blood cells in PD versus control blood samples [50]. To fully explore whether and how DJ-1 might act as a biomarker of PD and progression, more careful consideration of its various isoforms including posttranslational modifications is needed.
β-Amyloid and tau
β-Amyloid (Aβ) and tau proteins have been under intensive scrutiny in AD. Specifically, the 42 aa isoform of β-amyloid (Aβ1–42) and total (t-tau) and hyperphosphorylated tau (p-tau) concentrations in the CSF have recently been included in diagnostic criteria, and may be of use not only in the diagnosis of AD with dementia but also in predicting AD [51]. Parkinson’s disease ventricular CSF from post-mortem cases [52] showed that the p-tau-181:Aβ1–42 ratio and ApoA-1 levels were significantly associated with PD versus controls, demonstrating that AD markers may also be of use in PD. Reduced CSF Aβ1–42 levels have been suggested to be an independent predictor of cognitive decline in PD patients [53]. A combination of proteins examined by Shi et al. [37] found that the ratio of fractalkine:Aβ1–42 in CSF samples not only correlated with disease severity in a cross-sectional cohort of PD but also with disease progression in longitudinal samples. A recent study of CSF samples from the PPMI cohort described lower Aβ1–42 and p-tau associated with the diagnosis of PD, and also decreased t-tau levels associated with increased motor severity [35]. Differences between tremor-predominant and postural instability and gait disorder (PIGD) subtypes of PD suggested a potential association with prognosis: lower Aβ1–42 and p-tau were both associated with PIGD and not with the tremor-predominant phenotype of PD. Both PD severity and cognitive impairment/dementia in another study associated with the CSF complement 3:Aβ1–42 ratio and factor H:Aβ1–42 ratio [54], again supporting the usefulness of Aβ1–42. In 22 nondemented individuals with PD, CSF levels of Aβ1–42, t-tau and Aβ1–42:total tau correlated with various scores of cognition, including Logical Memory (delayed), Category Fluency, Digit Symbol, and Trails B minus A [55]. Recently, analysis of the DATATOP CSF samples from 403 participants measured t-tau, p-tau and Aβ1–42. In early PD, Aβ1–42 levels were found to have a weak negative correlation with total Unified Parkinson’s Disease Rating Scale (UPDRS) scores, and baseline p-tau:t-tau and p-tau:Aβ1–42 ratios were found to have a negative correlation with the rate of UPDRS change. While medications (selegiline and/or tocopherol) did not appear to alter biomarkers appreciably, a weak but significant positive correlation between the rate of change in t-tau level or t-tau:Aβ1–42 ratio and the change in UPDRS score was observed.
Antibodies targeting Aβ in AD are now under intensive study. If successful, repurposing such treatments from AD to PD will need to be considered. In this situation, prior experience with CSF protein biomarkers in AD will be invaluable, including help in anticipating potential pitfalls. In a phase 2 clinical trial of the anti-Aβ monoclonal antibody bapineuzumab in mild to moderate AD, reduced CSF t-tau and p-tau concentrations were associated with the study intervention, possibly reflecting an effect on the disease process [42]. Unfortunately, a phase 3 clinical trial detected no clinical benefit [43]. Solanezumab, an anti-Aβ antibody, was also recently tested in phase 2 and phase 3 randomized, double-blind, placebo-controlled trials in mild to moderate AD [44, 45]. The CSF unbound Aβ1–42 increased while the CSF unbound Aβ1–40 decreased in both phase 2 and phase 3 trials, suggesting a change in equilibrium in Aβ in the central compartment. The investigators interpreted the increase in CSF unbound Aβ1–42 as indicating a shift in “equilibrium” as the plaque was modified, yet phase 3 trials failed to meet their clinical primary endpoints, with no demonstration of significant cognitive improvement or functional improvement [45]. This echoes the situation in the CALM-PD, REAL-PET and ELLDOPA trials in PD described in above, in which biomarker changes did not reflect clinical outcomes. It remains to be determined whether the changes measured are truly irrelevant to clinical outcome, or whether the discordance arose from clinical aspects of the trial design. The CSF biomarker results in these AD studies nonetheless supported a central effect. With the possibility that intervention was made too late in the disease process, testing of solanezumab in an earlier “at-risk” population is now being pursued.
Markers of oxidative stress
Increased levels of oxidative stress contribute to PD pathogenesis, and not only are mitochondria the major source of cellular free radicals but defects in their biogenesis are now well documented in PD [56, 57]. Oxidative stress markers, including markers of redox status and oxidative damage, have therefore been of great interest in studies of various blood components and CSF in PD. There are many potential species that have demonstrated initial promise. Glutathione is a potent antioxidant, and an increase in its oxidized form was reported in plasma in PD versus control subjects [58]. Similarly for coenzyme Q10 (CoQ10), another potent antioxidant, two studies found its redox ratio in blood samples to be decreased in PD versus controls [59, 60], and in CSF the oxidized:total CoQ10 ratio was increased in PD [61]. Malondialdehyde is a product formed as reactive oxygen species degrade polyunsaturated lipids, and two studies have found significantly increased plasma levels of malondialdehyde associated with PD [58, 62]. Other markers of oxidative damage to lipids may be similarly elevated. In a study of PD compared with healthy controls, plasma F2-isoprostanes (a fatty acid peroxidation product) and cholesterol oxidation products were elevated in PD [63]. Oxidative damage to DNA has been recognized in plasma and CSF in PD, and notably 8-hydroxy-2 deoxyguanosine (8-OHdG), formed by direct oxidative damage to nucleosides, is elevated in plasma and urine in PD [64, 65].
None of these markers has yet been linked to PD progression, and therefore they are not helpful at this point in tracking the effect of potential neuroprotectants on disease progression. However, in clinical trials, they have the potential to determine the effects of an intervention on redox status in blood or CSF. This is important in understanding why (or sometimes more importantly, why not) a given drug may be effective. There are now a number of potential therapeutic agents acting on the interrelated pathways of mitochondrial dysfunction and oxidative stress [66], making this an ideal area to incorporate biomarkers for target engagement into clinical trials. With the recent lack of benefit in a large phase 3 clinical trial of high-dose CoQ10, inclusion of such markers might have proved informative. A recent study, however, suggested that findings may be more complex than anticipated. In an open-label, dose-escalation clinical trial testing CoQ10 up to 2400 mg daily in 16 individuals with early PD, plasma F2 isoprostanes and serum phospholipase A2 were reduced at CoQ10 doses of 400–1200 mg daily (as expected) but were actually increased at a dose of 2400 mg daily [67]. This serves to illustrate the potential complexities in translating preclinical promise into a clinically useful intervention.
Urate and other purine pathway compounds
Urate is a potent antioxidant, a metal chelator capable of binding iron, and in several rodent models of PD is neuroprotective [68–70]. In humans, it is the end product of the metabolic pathway for purines, and is subject to dietary influences. Independent studies have now detected decreased urate concentration in serum (for example [71]) and in CSF [72] in strong association with PD in men (although less so in women). Moreover, higher serum and CSF urate concentrations in post-hoc analyses of two large clinical trials have been associated with slower PD progression [72, 73]. Urate is therefore a high priority for interventional testing as a potential neuroprotectant in PD. Urate itself is rapidly degraded by intestinal bacteria after oral ingestion. However, its precursor, inosine (Figure 24.3), may be orally administered and has been shown to elevate serum urate. Recently, a phase 2 clinical trial of inosine was undertaken in individuals with de novo PD. Primary endpoints were safety and tolerability but also included a primary endpoint of ability to elevate serum and CSF urate concentration, i.e. as a means of testing “target engagement.” In this study, serum urate levels were elevated in the mild and moderate inosine dose treatment arms by 2 weeks, and were back to baseline at 1 month after cessation of the study drug. A single measurement of CSF urate levels found elevations in the mild (40%) and moderate (50%) inosine dose treatment arms in 44/75 participants from whom CSF was collected [74]. These findings therefore provide encouraging data upon which to proceed with larger-scale testing in which manipulated urate changes can be examined for correlation with clinical outcomes.
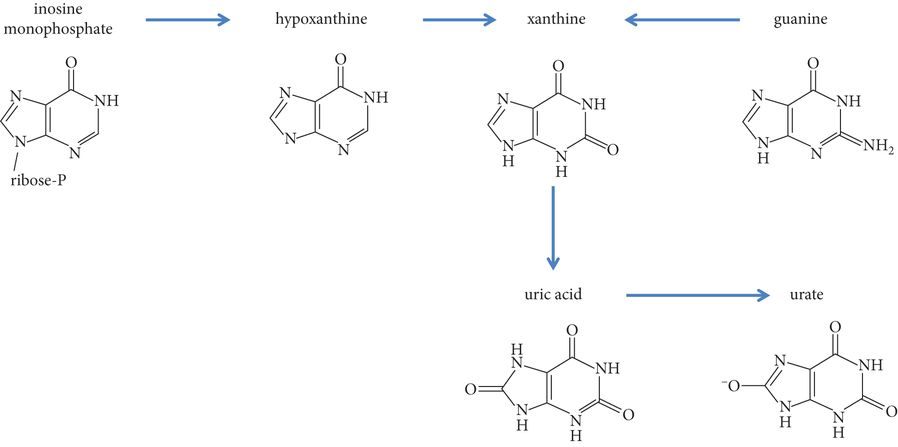
Metabolic pathway depicting the synthesis of urate from its precursor, inosine. Inosine has been tested in a phase 2 clinical trial with primary outcome measures of safety, tolerability and ability to elevate urate levels.
In addition to urate, altered purine metabolism (Figure 24.3) has been demonstrated in PD. Hypoxanthine concentration, and hypoxanthine:xanthine as well as xanthosine:xanthine ratios were found to be reduced in PD versus controls [75]. In a study of 217 unmedicated PD subjects compared with 26 control subjects, the mean xanthine:homovanillic acid ratio was significantly higher in PD subjects, and this ratio was further elevated at the 24-month repeat testing, raising the possibility that the degree of abnormality of the xanthine:homovanillic acid ratio might reflect disease progression [14]. Further longitudinal studies are now warranted, as this seems to be a promising pathway in studying PD progression.
Markers of inflammation
Inflammatory pathways are now thought to play a significant role in PD pathogenesis. Microglial activation has been well documented in autopsy specimens in PD. Moreover, neuroimaging using the peripheral benzodiazepine receptor ligand PK11195 supports ongoing microglial activation during a patient’s lifetime [57]. Although studying inflammatory markers has not yet revealed candidates for use in delineating the progression of motor PD, it is possible that such markers will be useful in demonstrating target engagement and pathway effects for future anti-inflammatory intervention. Several studies have now described disrupted inflammatory pathways in blood and CSF, although many of these findings remain to be replicated.
Interleukin (IL)-10 and the chemokine RANTES (Regulated on Activation, Normal T-cell Expressed and Secreted) have been found at increased levels in serum in PD compared with controls [76]. Decreased mannan-binding lectin levels and elevated tumor necrosis factor (TNF)-α levels have been detected in association with PD [77]. In addition to this finding in plasma, an increase in TNF-α has been reported in CSF in patients with PD versus controls [78]. One interesting possibility is that nonmotor symptoms may be associated with a pro-inflammatory state [79]. In one study, not only PD severity but also cognitive impairment and dementia were associated with the CSF complement 3:Aβ1–42 ratio and factor H:Aβ1–42 ratio [54]. Increased plasma levels of the soluble TNF receptors TNFR1 and TNFR2 were associated in one study with worse scores on psychometric testing [80]. In a study of CSF from 87 PD patients and 33 controls, measures of inflammatory markers including C-reactive protein, IL-6, TNF-α, interferon γ-induced protein-10, monocyte chemotactic protein-1 and macrophage inflammatory protein 1-β did not differ between groups, but instead inflammatory markers were significantly associated with worse measures of depression, anxiety, fatigue and cognition within the PD group [81]. These independent findings therefore suggest a potential area of focus upon cognitive dysfunction in PD that warrants further study.
Perturbations in cellular components of the immune system have been documented. A higher population of circulating CD4-bright+/CD8-dull+ lymphocytes (usually seen in the thymus) has been demonstrated in PD compared with control subjects [82]. Decreased numbers of CD4+CD45RA+ (naive) T cells and increased numbers of CD4+CD45RO+ (memory) T cells and TCRγδ+ cells have also been observed [83], and this same study found increased HLA-DR expression on CSF monocytes. Whether these will provide benefit as PD biomarkers remains to be seen.
Abnormal antibody expression has also been detected in PD. Increased levels of anti-melanin antibodies detected by ELISA were observed in one study in serum from individuals with PD versus age-matched controls, and levels were inversely correlated with PD duration [84]. Of particular interest, however, are two examples of studies in which antibody measures were incorporated into clinical trials [85]. The CSF was examined for antineuronal antibodies in clinical trial participants who had undergone adrenal medulla cell transplantation as a potential treatment for PD, and transplantation was found to affect the presence of these antibodies. The measurement of anti-glutamic acid decarboxylase (GAD) antibodies also serves to illustrate a potential use of biomarkers in clinical trials – that of detecting potentially undesirable outcomes. When the first human phase 1 study of GAD gene therapy was performed in PD, a potential concern was that a “host” immune response might be mounted. Serum samples from participants with PD in this open-label trial were therefore tested for the presence of neutralizing antibodies. No induction of such antibodies was observed over the 1-year clinical trial period [86]. Although these are two isolated examples, it should stimulate more thoughtful inclusion of antibody measures in future clinical trials that involve the introduction of a “foreign” biological material. Recently, the existence of anti-α-synuclein antibodies has also been associated with PD [87, 88].
Untargeted approaches: the “omics”
In contrast to the targeted markers discussed above, the “omics” approach makes use of emerging technologies and bioinformatics for unbiased and systematic evaluation of patterns of variations in RNAs (transcriptomics), proteins (proteomics) or small molecules (metabolomics), and has been highly promising as a means to develop disease biomarkers in neurodegenerative diseases including PD [89]. Sophisticated statistical methods allow large amounts of data to be organized for data mining and informatics, making these techniques potentially extremely powerful. In general, “omics” offers the possibility for identification of one or more species (protein, peptide, RNA or small molecule) or a composite “fingerprint” that could potentially reflect disease progression and/or response to a specific treatment. Although early in application to PD, those studies that have successfully identified potential PD biomarkers are outlined below. Most of the findings described still require independent validation, are cross-sectional and are focused on markers that differ between PD and healthy or disease controls. Longitudinal changes and changes in response to treatment remain to be investigated in the majority of cases. At the present time, “omics” studies remain highly technically challenging.
Transcriptomics
The transcriptome comprises all mRNAs present in a given sample and therefore reflects both transcription from DNA and post-transcriptional regulation. Transcript patterns are determined using high-throughput microarray techniques, and multiple studies have demonstrated disruption of gene expression in PD brain tissue [90–92], providing hope that such changes may be reflected in the CSF and/or blood. There are now multiple studies that support this possibility. Grunblatt et al. [91] selected 12 candidate transcripts upon differential expression in brain tissue, and used quantitative reverse transcription PCR to demonstrate that the combination of transcripts (encoding proteasome subunit-α type 2, laminin β-2, aldehyde dehydrogenase 1 family-member A1 and histone cluster-1 H3e) in blood achieved a sensitivity and specificity of over 80% for association with PD. A case–control study of 66 subjects similarly defined a combination of eight transcripts whose levels were associated with risk of PD, and these results were then validated in a smaller set of independent samples [94]. Many transcripts identified in such studies fit into known pathways: for example, in a study of 22 mRNAs found to differ between PD and controls, the co-chaperone ST13 stands out as a stabilizer of heat-shock protein 70, known to modify α-synuclein misfolding and toxicity, among numerous other roles. Differences are evident in early PD: for example, transcriptomic analysis of blood samples identified altered levels in p19 S-phase kinase-associated protein 1A, huntingtin-interacting protein-2, aldehyde dehydrogenase family 1 subfamily A1, 19S proteasomal protein PSMC4 and heat-shock 70 kDa protein 8 [95]. A panel of these transcripts had high sensitivity and specificity in identifying PD cases, was validated in advanced PD and was unaffected by medication status. Regarding the utility of defining a response to treatment, although the above study found no difference according to PD medication, other studies have found a number of effects of antidepressant and antipsychotic medications on transcripts with potential effects in PD [96]. Lithium and bupropion, for example, downregulate brain-derived neurotrophic factor (BDNF) transcripts, and amitriptyline upregulates the dopamine-receptor D2 (DRD2) and DRD3 genes. These provide a proof of principle that changes in the transcriptome might be sensitive and informative to treatment interventions. Perhaps defining transcriptome responses to potential neuroprotectants might be informative in future studies as a measure of target pathway engagement.
Proteomics
Proteomic analysis involves separation of the protein or peptide constellation of a given biosample, followed by their identification by mass spectrometry-based techniques and advanced bioinformatics to determine species or combinations of species that may be associated with a specific state. The first proteomics study in PD used two-dimensional gel electrophoresis in substantia nigra pars compacta extracts to identify a set of nine proteins that were differentially expressed in PD compared with healthy controls [97], and further analysis increased this number to 221 [98]. The study is encouraging in identifying proteins likely to be involved in PD pathogenesis, such as glutathione S-transferase and other genes involved in glutathione metabolism, and suggests that these are worthy of more targeted studies. In a separate study by Abdi et al. [99], over 1500 CSF proteins were analyzed using isobaric tagging for relative and absolute protein quantification (iTRAQ), followed by mass spectrometry in samples from individuals with PD (clinical diagnosis), DLB (autopsy confirmed), AD (autopsy confirmed) and controls. Seventy-two of these manifested altered expression in PD. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) has also been used to identify a set of proteins that are differentially expressed between PD and controls [1, 100, 101]. Furthermore, recent analysis of proteins captured by magnetic bead-based weak cation exchange followed by MALDI-TOF MS identified a combination of five biomarkers discriminating PD and healthy controls with high sensitivity (85%) and specificity (70%) in a blinded analysis [102]. Other approaches, including two-dimensional gel electrophoresis and MS, are also highly encouraging that differentiation of PD is possible on the basis of such proteomic fingerprints [103, 104].
These potential markers have yet to be examined in longitudinal cohorts, and how they respond to PD treatment is unknown. However, a recent proteomic analysis of CSF obtained from a small number of subjects before and after deep-brain stimulation therapy for PD involved two-dimensional differential gel electrophoresis, in combination with MALDI-TOF and tandem TOF MS, or electrospray ionization MS. The investigators determined that two proteins, extracellular superoxide dismutase (SOD) and tetranectin, were significantly elevated in samples taken after DBS [105]. In one patient, a further CSF sample was analyzed with the stimulator present but turned off, and it was found that extracellular SOD and tetranectin had now declined. The authors suggested that changes were therefore due to stimulation. This is a highly preliminary observation but nonetheless suggests that proteomic analysis will be sensitive enough to detect at least some treatment-related changes.
Metabolomics
Metabolomic analysis focuses on the array of low-molecular-weight species in a given sample, usually CSF or plasma. Metabolomic profiling using plasma samples has now been used to successfully distinguish subjects with PD versus healthy controls using high-performance liquid chromatography with electrochemical coulometric array detection to quantitatively assess approximately 2000 small analytes [64]. In a small number of samples (PD nonmedicated: n= 15; control: n= 25) metabolomics analysis was able to completely distinguish the two groups (P < 0.01). Using this same platform, it was demonstrated that individuals with idiopathic PD and leucine-rich repeat kinase 2 (LRRK2)-associated PD could also be distinguished from each other and from control subjects on the basis of their metabolomics profile [75]. As discussed above, when examining levels of individual analytes, urate was found to be lower in idiopathic and LRRK2 PD than in control samples [64, 75]. A smaller, nonstatistically significant decrease was measured in LRRK2 nonmanifesting carriers [75], leaving the question as to whether this would be a helpful premotor-phase tool unanswered. Other specific purine metabolites (hypoxanthine, HVA:xanthine ratio, xanthosine:xanthine ratio) have been demonstrated to be significantly reduced in a small number of unmedicated PD patients versus controls [75]. Although most of these metabolomics analyses did not involve a longitudinal component, in one study, as noted earlier, metabolomic analysis was instrumental in detecting HVA:xanthine ratio abnormalities in PD that changed with disease progression over a 2-year period [13, 14].
Related posts:
 Oral dopamine agonists in the management of Parkinson’s disease
Management of disease-related behavioral disturbances in Parkinson’s disease
A neurobehavioralist approach to the management of cognitive impairment in Parkinson’s disease
Lessons learned: symptomatic trials in early Parkinson’s disease
Controversy: midstage vs advanced-stage deep-brain stimulation in the management of Parkinson’s disease
Lessons and challenges of trials for other nonmotor complications of Parkinson’s disease
Oral dopamine agonists in the management of Parkinson’s disease
Management of disease-related behavioral disturbances in Parkinson’s disease
A neurobehavioralist approach to the management of cognitive impairment in Parkinson’s disease
Lessons learned: symptomatic trials in early Parkinson’s disease
Controversy: midstage vs advanced-stage deep-brain stimulation in the management of Parkinson’s disease
Lessons and challenges of trials for other nonmotor complications of Parkinson’s disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


