Fig. 1
Histogram of documented cerebral vasospasm. The greatest frequency of vasospasm was during days 11–14 (5 of 7 patients)
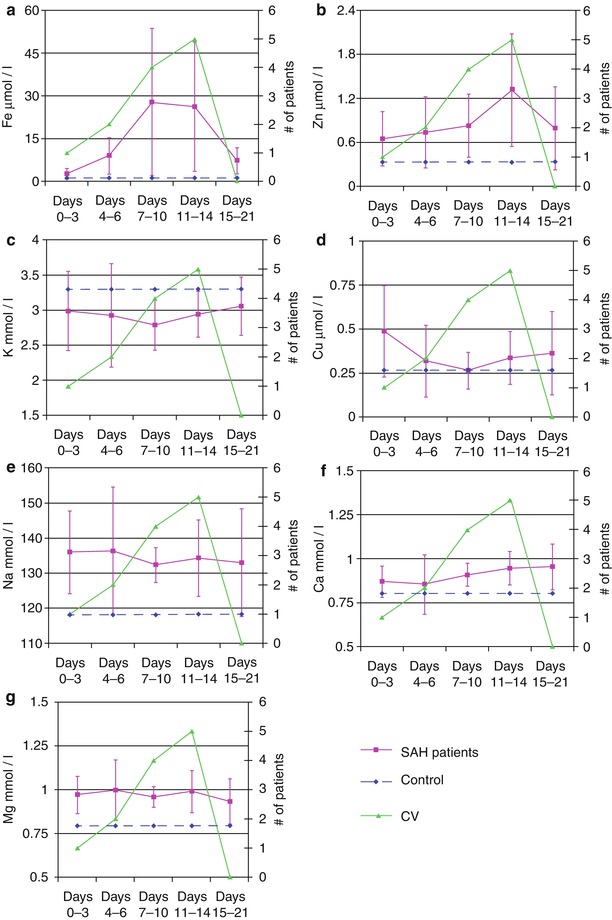
Fig. 2
Changes in the metal cation concentration in the cerebrospinal fluid of subarachnoid hemorrhage patients. The horizontal dashed line is the concentration of the metal cation in the control group (this was not collected over time and is plotted as a line for reference). The number of patients who experienced cerebral vasospasm during a given time period is plotted on the secondary axis. (a) Fe2+, (b) Zn2+, (c) K+, (d) Cu2+, (e) Na+, (f) Ca2+, (g) Mg2+
Serum Chemistry Values
The serum chemistry values of Na+, K+, Ca2+, and Mg2+ were assessed until PHD 21. The average concentration of Na+ and K+ exhibited no deviation from the normal range throughout the entire time frame (Fig. 3a, b). However, there were significant fluctuations within these limits. The initial serum level for K+ was significantly lower than PHD 7–10 (P = 0.048). The serum level of Na during PHD 7–10 was significantly lower than PHD 0–3 (P = 0.002) and PHD 4–6 (P = 0.009). Mg2+ levels were above normal through PHD 14; peaking during PHD 4–6 (1.34 mmol/L) (Fig. 3c). The peak was significantly higher than the level recorded during PHD 15–21 (P = 0.024). Lastly, Ca2+ levels were below normal through PHD 7, with the lowest concentration occurring during the first time period (8.0 mg/dL). Ca2+ levels returned to normal for PHD 7 through PHD 21 (Fig. 3d). The peak level during PHD 15–21 was significantly higher than during PHD 0–3 (P = 0.013), PHD 4–6 (P = 0.003), and PHD 11–14 (P = 0.024).
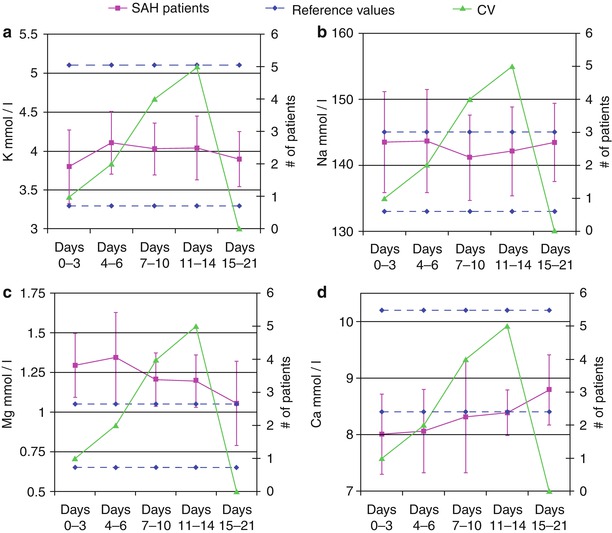
Fig. 3
Changes in the serum chemistry values for subarachnoid hemorrhage patients. The horizontal dashed lines represent the high and low limits of the normal range. The number of patients who experienced cerebral vasospasm during a given time period is plotted on the secondary axis. (a) K+, (b) Na+, (c) Mg2+, (d) Ca2+
Discussion
Blood products in the CSF after SAH are attributed to CV and early brain injury, but the mechanism remains elusive. The time between SAH and CV provides a unique window for therapeutic intervention. Proposed pathophysiological mechanisms of CV and early brain injury have included oxygen free radical (OFR) release from the degradation of hemoglobin in the CSF, an inflammatory response from blood products in the subarachnoid space, and lower levels of vasodilators such as carbon monoxide (CO) released by the degradation of hemoglobin by the enzyme, heme-oxygenase (HO)-1 [4, 9, 10, 26].
Knowledge of the metal ion composition of CSF and its association with CV may provide insight into the pathways that trigger vasospasm, thereby promoting the development of neuroprotective strategies. We demonstrated that two of the measured metal ions, Fe2+ and Zn2+, change significantly over time and parallel the time course of radiographic and clinical CV. Thus, both of these ions may have important roles in the process of CV and, in the case of Zn2+, may also be a marker of neuronal injury.
Changes in the CSF Fe2+ concentrations in this study paralleled the onset and resolution of CV seen in the SAH population. The red blood cells, released into the subarachnoid space after aneurysm rupture, lyse, releasing hemoglobin into the CSF. HO-1 degrades heme, releasing Fe2+ and the byproduct, CO. Fe2+ is responsible for a number of oxidative pathways that create OFRs, stimulating an inflammatory response as the iron is removed by the resident macrophage-like cells of the nervous system, called microglia [5, 27, 28, 30]. This inflammatory process can promote CV and neuron toxicity [15]. Interestingly, both Fe2+ and Zn2+ decreased during the final time period (PHD 15–21) of CV in parallel with the incidence of CV in this study population, further supporting their role in the pathophysiology of vasospasm.
Zinc CSF concentrations also paralleled the incidence of CV in this study, peaking just after those of Fe2+. Zn2+ has many key roles in cellular metabolism, protein synthesis, and injury pathways, such as OFR formation, ATP-depletion, transcription factors (the zinc finger), and the initiation of glycolysis. Each of these roles is potentially important in the development of neuronal injury and CV [22, 23, 25]. Another potentially interesting role of Zn2+ in CV is as a competitive inhibitor of HO-1 when in the form of Zn2+ protoporphyrin-IX, a hemoglobin analog found in red blood cells [9, 30]. HO-1 has a higher affinity for hemoglobin than its Zn2+ analog, but its competitive inhibition of HO-1 can lower carbon monoxide production, a vasodilating gas molecule, thereby potentiating CV [4, 9, 10, 24, 26, 30]. Zn2+ is also involved with endothelial and smooth muscle cell ultrastructural composition and smooth muscle relaxation, each an important concept when considering the potential role of Zn2+ in CV pathways [20–22].
Whether Zn2+ is directly involved with CV as discussed above or represents a marker of neuronal injury is an important question. “Zinc neurons” have been described in the cortex. These neurons store Zn2+ in vesicles contained within the cell and have been shown to release Zn2+ during ischemia, a process called “zinc dumping” [11–14]. Thus, elevated Zn2+ levels may not only be seen because of its role in CV modulation, but also as a marker of a more subtle ischemic process and cellular injury from CV occurring at the neuronal level. Lastly, Zn2+ plays an important role in immune modulation. The resident immune cells of the nervous system, microglia, are activated from a scavenging mode that is more benign to a more activated cytotoxic phenotype that releases OFRs and promotes inflammation [14, 16, 18, 24]. This proinflammatory role of Zn2+ may be an important target for antispasmotic therapy and neuroprotection in aneurysm SAH patients.
Although the concentrations of the cations Na+, Cu2+, Mg2+, and Ca2+ in the CSF did not change significantly during the course of CV, they were different from control values. This suggests that these metal ions likely reflect contamination of CSF space with serum plasma that entered the subarachnoid space during the initial aneurysm rupture, release of cytosolic contents from blood cells, and changes in the serum levels over the course of SAH treatment. The latter is particularly true for Na+ and Mg2+ where patients are routinely placed on magnesium sulfate infusions intravenously for neuroprotection and, in selected cases, hypertonic 3 % sodium chloride solutions to maintain mild hypernatremia to reduce brain tissue swelling.
Serum Na+ and K+ concentrations were significantly lower early after SAH. Previous reports have demonstrated similar results and attribute this to cortisol release during the stress response [29]. Cortisol causes Na+ and K+ to be lost from the kidneys and, additionally, the K+ to be taken in by cells from the extracellular space. This may explain the initial lower levels of these metal ions. Additionally, it is well known that the hypothalamic–pituitary axis malfunctions after SAH, seen clinically as the syndromes of inappropriate antidiuretic hormone secretion and cerebral salt wasting [8]; both causing hyponatremia. Serum magnesium levels are elevated above control levels in this study because magnesium sulfate continuous infusions are initiated as part of our standard of care because of data demonstrating its action as a neuroprotectant. Clinically, we aim to keep serum Mg2+ levels at 2.5–3.5 mEq/L. Lastly, serum Ca2+ levels were lowest early after SAH. It is uncertain whether this hypocalcemia is related to the stress response, the routine use of a calcium channel blocker (Nimotop) and MgSO4 for neuroprotection, or some other undefined etiology [3]. Further evaluation of trends in serum Na+, K+, and Ca2+ levels in the setting of SAH seem warranted. Additionally, future studies looking at trends in serum Mg2+ concentrations in patients without magnesium sulfate infusions would be interesting to see whether there are any changes that occur over the course of CV, because smaller changes may not be measured when the patients are being administered Mg2+ infusions.
Related posts:
 Role of Erythropoietin in Aneurysmal Subarachnoid Haemorrhage: From Bench to Bedside
Modulation of Spreading Depolarizations
Care of Aneurysmal Subarachnoid Hemorrhage: State of the Art
Rate in the Sylvian Cistern Is Associated with the Severity of Cerebral Vasospasm After Subarachnoid Hemorrhage
Role of Erythropoietin in Aneurysmal Subarachnoid Haemorrhage: From Bench to Bedside
Modulation of Spreading Depolarizations
Care of Aneurysmal Subarachnoid Hemorrhage: State of the Art
Rate in the Sylvian Cistern Is Associated with the Severity of Cerebral Vasospasm After Subarachnoid Hemorrhage
 Changes in Brain Tissue Oxygen During Endovascular Treatment of Cerebral Vasospasm
Changes in Brain Tissue Oxygen During Endovascular Treatment of Cerebral Vasospasm
 Secondary Injury After Subarachnoid Hemorrhage in Light of Multimodal Advanced Neuroimaging, Intracranial and Transcranial Neuromonitoring: Beyond Vasospasm
Secondary Injury After Subarachnoid Hemorrhage in Light of Multimodal Advanced Neuroimaging, Intracranial and Transcranial Neuromonitoring: Beyond Vasospasm
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


