Neurodegenerative disorder
System involved
Neurologic/psychiatric/behavioral abnormalities
Inheritance pattern
Infantile Refsum’s disease
Peroxisomal disorders
Rapid, jerky eye movements (nystagmus); progressive muscle weakness and wasting; poor balance and coordination (ataxia); hearing loss
Austosomal recessive
Zellweger syndrome
Peroxisomal disorders
Weak muscle tone (hypotonia), feeding problems, hearing loss, vision loss, and seizures
Austosomal recessive
Neonatal adrenoleukodystrophy
Peroxisomal disorders
Hypotonia, vision problems, hearing loss, liver dysfunction, developmental delay, intellectual disability
Austosomal recessive
Ornithine transcarbamoylase deficiency
Urea cycle disorders
Feeding difficulties, lethargy, and respiratory distress, irritability, temper tantrums, inconsolable crying, ataxia, seizures, hyperactivity
X-linked recessive
Argininosuccinicaciduria
Urea cycle disorders
Feeding difficulties, lethargy, mental retardation, recurrent generalized convulsions, ataxia, poorly controlled breathing rate or body temperature
Austosomal recessive
Arginosuccinate lyase deficiency
Urea cycle disorders
Lethargy, poor feeding, neurocognitive deficiencies (attention deficit hyperactivity disorder) [ADHD], developmental disability, seizures, and learning disability
Austosomal recessive
Pyruvate carboxylase deficiency
Mitochondrial disorders
Weak muscle tone (hypotonia), abnormal movements, seizures, and coma
Autosomal recessive
Glutaricaciduria type II deficiency
Mitochondrial disorders
Poor feeding and decreased activity, and vomiting
Autosomal recessive
Carnitine palmitoyltransferase deficiency type II
Mitochondrial disorders
Seizures, cardiomyopathy, respiratory failure, seizures, liver failure, cardiomyopathy, and an irregular heartbeat (arrhythmia)
Autosomal recessive
Maple syrup urine disease
Amino acid and organic acid disorders
Intermittent periods of ataxia, drowsiness, behaviour disturbances, and seizures
Autosomal recessive
Nonketotic hyperglycemia
Amino acid and organic acid disorders
Lethargy, profound hypotonia, intractable generalized or myoclonic seizures, apnea, feeding difficulties
Autosomal recessive
Methylmalonic acidemia
Amino acid and organic acid disorders
Hypotonia, lethargy, recurrent vomiting, profound metabolic acidosis, spastic quadriparesis, dystonia, and severe developmental delay
Autosomal recessive
Menkes kinky hair syndrome
Copper transport system
Weak muscle tone (hypotonia), sagging facial features, seizures, developmental delay, and intellectual disability
X linked recessive
Spinal muscular atrophy type 1
Peripheral nerves
Muscle weakness, respiratory failure, a weak cry; problems feeding; recurrent episodes of pneumonia, excessive sweating (hyperhidrosis), loss of bladder and bowel control, and an irregular heartbeat (arrhythmia)
Autosomal recessive
Smith-Lemli-Opitz syndrome
Cholesterol metabolism
Microcephaly, moderate-severe intellectual disability, Autistic behaviours, hyperactivity, aggressiveness and self injurious behaviours weak muscle tone (hypotonia), feeding difficulties, sleep cycle disturbance
Autosomal recessive
Galactosemia type I
Carbohydrate metabolism
Poor feeding, delayed development, clouding of the lens of the eye (cataract), speech difficulties, and intellectual disability
Autosomal recessive
Table 11.2
Some neurodegenerative disorders in infancy showing the principal systems affected, neurologic/psychiatric/behavioral abnormalities and their inheritance patterns
Neurodegenerative disorder | System involved | Neurologic/psychiatric/behavioral abnormalities | Inheritance pattern |
|---|---|---|---|
Farber’s disease | Lysosomal storage diseases | Paralysis of the arms and legs (quadriplegia), seizures, loss of speech, involuntary muscle jerks (myoclonus), and developmental delay | Autosomal recessive |
Krabbe’s disease | Lysosomal storage diseases | Irritability, muscle weakness, feeding difficulties, episodes of fever without any sign of infection, stiff posture, and slowed mental and physical development, vision loss and seizures | Autosomal recessive |
Tay-Sachs disease | Lysosomal storage diseases | Loss of motor skills such as turning over, sitting, and crawling exaggerated startle reaction to loud noises, seizures, vision and hearing loss, intellectual disability, and paralysis | Autosomal recessive |
Alexander disease | Leukodystrophies | Megalencephaly, seizures, spasticity, intellectual disability and developmental delay | Autosomal dominant |
Canavan disease | Leukodystrophies | Feeding and swallowing difficulties, seizures, and sleep disturbances, developmental delay, hypotonia, macrocephaly, abnormal posture, and intellectual disability | Autosomal recessive |
Pelizaeus-Merzbacher disease | Leukodystrophies | Impaired intellectual functions, such as language and memory, delayed motor skills, such as coordination and walking | X linked recessive |
Leigh disease | Mitochondrial disease | Hypotonia, involuntary muscle contractions (dystonia), ataxia, peripheral neuropathy, weakness or paralysis of the muscles that move the eyes (ophthalmoparesis); rapid, involuntary eye movements (nystagmus); or degeneration of the nerves that carry information from the eyes to the brain (optic atrophy), severe breathing problems, and hypertrophic cardiomyopathy | Autosomal recessive/X linked recessive/Sporadic |
Medium-chain acyl CoA dehydrogenase deficiency | Mitochondrial disease | Lethargy, vomiting, seizures, breathing difficulties, liver problems, coma, and hypoglycemia | Autosomal recessive |
Phenylketonuria | Amino acids and organic acid diseases | Profound intellectual disability, developmental delay, microcephaly, seizures (e.g., tonic-clonic, myoclonic, infantile spasms), tremors, athetosis, and spasticity, autistic behavior and attention-deficit–hyperactivity disorder | Autosomal recessive |
Glutaric aciduria type I | Amino acids and organic acid diseases | Macrocephaly, muscular spasms, jerking, rigidity, or decreased muscle tone | Autosomal recessive |
Lowe syndrome | Central nervous system and eyes and kidneys | Delayed development, impaired vision, moderate to severe intellectual disability, Kidney abnormalities, seizures, weak muscle tone from birth (neonatal hypotonia), feeding difficulties and problems with breathing | X linked recessive |
Biotinidase deficiency | Enzymes that depend on Vitamin biotin | Seizures, weak muscle tone (hypotonia), breathing problems, and delayed development, hearing loss, eye abnormalities, loss of vision, problems with movement and balance (ataxia), skin rashes, hair loss (alopecia) | Autosomal recessive |
Table 11.3
Some neurodegenerative disorders in childhood and adolescence showing the principal systems affected, neurologic/psychiatric/behavioral abnormalities and their inheritance patterns
Neurodegenerative disorder | System involved | Neurologic/psychiatric/behavioral abnormalities | Inheritance pattern |
|---|---|---|---|
Charcot Marie tooth disease | Peripheral nerves | Balance difficulties, clumsiness, and muscle weakness in the feet, loss of sensation and wasting (atrophy) of muscles in the feet, legs, and hands | Autosomal dominant |
Wolfram syndrome (diabetes insipidus, diabetes mellitus, optic atrophy and deafness) [DIDMOAD] | Multisystem | Depression, paranoia, auditory or visual hallucinations, violent behavior, dementia, suicide | Autosomal recessive |
Symptomatic progressive myoclonic epilepsies (such as Unverricht-Lundborg disease (ULD) and Lafora disease) | Central nervous system and for Lafora disease in addition-affects heart, liver and muscle | Myoclonic jerks and tonic-clonic seizures visual hallucinations (occipital seizures), progressive neurologic degeneration including cognitive and/or behavioral deterioration, dysarthria, and ataxia | Autosomal recessive |
Metachromatic leukodystrophy | Central and peripheral nervous system | Anxiety, depression, emotional lability, social withdrawal, schizophrenia, poor memory | Autosomal recessive |
Late-onset GM2 gangliosidosis | Lysosomal disorder | Dystonia, intention tremor, dementia, obsessional paranoia, hallucinations, acute psychosis, dysarthria | Autosomal recessive |
Juvenile Huntington’s disease | Basal ganglia | Rapid, significant drop in overall school performance, depression, gait disturbances, tremors or slight involuntary movements, seizures, obsessive compulsive disorder, mania, sexual inhibition or inappropriate sexual behaviours | Autosomal dominant |
Fabry’s disease | Lysosomal storage disorder | Pain, particularly in the hands and feet (acroparesthesias); a decreased ability to sweat (hypohidrosis); cloudiness of the front part of the eye (corneal opacity); ringing in the ears (tinnitus); and hearing loss | X-linked recessive |
Kearns-Sayre syndrome | Mitochondrial disease | Cardiac conduction defects, ataxia, muscle weakness, deafness, kidney problems, and a deterioration of cognitive functions (dementia) | Variable |
MELAS syndrome, NARP | Mitochondrial disease | Recurrent stroke-like episodes in the brain, migraine-type headaches, vomiting and seizures, general muscle weakness, exercise intolerance, hearing loss | Variable |
MERRF syndrome | Mitochondrial disease | Myoclonus (muscle jerks), seizures, ataxia, muscle weakness, hearing impairment | Variable |
Juvenile neuronal ceroid lipofuscinosis | Lysosomal storage disorder | Inappropriate behavior, thought disorder, paranoia with hallucinations, delusions | Autosomal recessive |
Subacute sclerosing panencephalitis | Central nervous system | Deterioration in learning or schoolwork, involuntary movements, deterioration in the thought processes, myoclonic jerks, epileptic seizures may or may not occur | Sporadic |
Adrenoleukodystrophy | Central nervous system and the adrenal glands | Difficulty reading or writing, obsessional behavior, irritability, social withdrawal, dementia | X-linked recessive |
Variant Creutzfeldt–Jakob disease | Central nervous system | Dementia, blurred vision, gait changes, disorientation, hallucinations, lack of coordination, myoclonic jerks or seizures, personality changes, sleepiness, speech impairment | Unknown |
Wilson disease | Multisystem especially liver, brain, and eyes | Antisocial behavior, anxiety, depression, manic depressive psychosis, clumsiness, tremors, difficulty walking and mood swings | Autosomal recessive |
Multiple sclerosis | Central and peripheral nervous system | Overwhelming fatigue, visual disturbances, altered sensation and difficulties with mobility, muscle stiffness (spasticity), exaggerated reflexes (hyperreflexia), or poor bladder control | Unknown |
Friederich’s ataxia | Central and peripheral nervous system -spinocerebellar degeneration | Ataxia, gradual loss of strength and sensation in the arms and legs, muscle stiffness (spasticity), impaired speech, hypertrophic cardiomyopathy, impaired vision, hearing loss, or an abnormal curvature of the spine (scoliosis) | Autosomal recessive |
Gaucher’s disease type III | Lysosomal storage disorder | Hepatosplenomegaly, Anemia, Thrombocytopenia, Bone disease (bone pain and fractures), seizures and slowing of horizontal eye movements | Autosomal recessive |
Regression and Progressive Neurological Deterioration Are Cardinal Features
The presentation of a child with a neurodegenerative developmental disorder has varied manifestations. Some children may present with a history of regression in previously acquired developmental milestones, or history of a delay in acquisition of milestones, history of no further attainment of milestones following previously normal progress or a history of total loss of previously acquired milestones. This is graphically illustrated in Fig. 11.1 that demonstrates the possible developmental trajectories of child development. Child (A) depicts the normally developing child who attains the neurodevelopmental milestones at the appropriate successive age. Child (B) shows a child having a fairly stable developmental progress however this is made at twice the successive age and is a picture typical of children with cognitive impairment. Child (C) shows an initial developmental progress which then levels off with hardly any more variation. This represents a child with a neurodegenerative disorder even in the absence of developmental regression. Child (D) initially exhibits a similar trend as is seen in child (C) nonetheless this is followed by gradual loss of developmental skills and is a picture that typifies “classic” neurodegenerative disorder. Child (E) on the other hand shows that this gradual loss of skills is not necessarily a smooth uninterrupted process but may proceed in intermittent bursts of deficits which may denote stress arising from the effects of injury or illness.
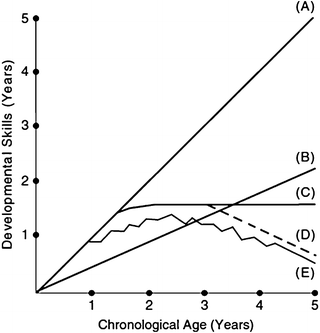
Fig. 11.1
Graphic representation of childhood development (Reproduced with permission from Goldstein E. M, & Holden K.R, (2009), Neurodegenerative Disorders: Variable Clinical Presentation of Cognitive Decline, in Bernard L. Maria (ed.), Current Management in Child Neurology (4th Edition, pp. 322–336), Shelton, CT: People’s Medical Publishing House, (PMPH-USA))
In certain circumstances it can be challenging to differentiate between a plateauing of skills, a pseudo-regression and real onset of a neurodegenerative condition in a child with a pre-existing neurological deficit. In such situations it is important to review the authenticity of the original diagnosis and institute the appropriate measures where possible to reverse the condition. Some causes of pseudo-regression may include: depression especially in adolescents, poorly controlled and subtle epileptic seizures as is seen in unrecognized absence seizures or non-convulsive (“subclinical”) status epilepticus, acquired hypothyroidism, substance abuse, lead encephalopathy, repeated traumatic brain injury and in children deprived of emotional contact as seen in prolonged hospitalizations [13] or in those who witness domestic violence [14].
Initial Evaluation of Children Suspected of Suffering from Neurodegenerative Disorders
A comprehensive history taking followed by detailed physical examination is paramount when evaluating children with suspected neurodegenerative disorders [4, 12, 15]. A thorough history taking is essential for the assessment of any child with developmental regression in order to exclude other disorders that may mimic neurodegenerative disorders. Following an uneventful pregnancy and delivery of a normal full term child, the child with suspected neurodegenerative disorder may either present with an acute and rapidly progressive or a vague and slowly progressive course. For the latter course it is crucial that it should be distinguished from a static non-progressive disorder. The slowly progressive course primarily affects the white matter and can commence in infancy, childhood or adolescence. Usually the acute onset predominantly affects the gray matter and is seen in neonatal or early infancy with the symptoms commencing a few days after birth.
Following an in-depth history, a formulation of differential diagnoses is made to come to a diagnostic hypothesis directing subsequent laboratory investigations. A classic distinction in the group of neurodegenerative disorders involves those with predominant white matter involvement (such as Multiple Sclerosis, Adrenoleucodystrophy, Alexander Disease, Canavan Disease, Krabbe Leukodystrophy, Metachromatic Leucodystrophy) versus those with predominant gray matter involvement (such as Mitochondrial Encephalopathies, Neuronal Ceroid Lipofuscinonis, Progressive Infantile Poliodystrophy, and the Symptomatic Progressive Myoclonic Epilepsies) [16].
White matter disorders typically feature onset in late childhood, with initially normal cognitive functions, but early and prominent spasticity and cerebellar signs, gait difficulties, and early peripheral neuropathy due to demyelination. Later signs occasionally include focal neurologic deficits, seizures and megalencephaly. Other signs are absence of exaggerated reflexes and optic atrophy. The electroencephalogram (EEG) shows diffuse delta slowing, the electromyogram (EMG) shows slowed nerve conduction velocity and evoked potentials are prolonged or absent.
Disorders primarily affecting white matter can be subdivided into two broad categories, demyelinating and dysmyelinating/hypomyelinating. Demyelination occurs when immune-mediated inflammatory responses, toxic exposure, or vascular injury destroys previously normal myelin, as for example in multiple sclerosis, acute disseminated encephalomyelitis, acute hemorrhagic leukoencephalitis and myelinoclastic diffuse sclerosis of Schilder. These white matter disorders likely have an immunological basis but their mechanisms are not completely understood. Disorders that involve dysmyelination or hypomyelination include metabolic diseases affecting the nervous system such as galactosemia, pyridoxine-dependent seizures, infantile Refsum’s Disease, metabolic disorders of lipid metabolism (e.g., Metachromatic Leukodystrophy, Krabbe’s Disease, and diseases affecting myelin proteins (e.g., Pelizaeus-Merzbacher) (see Tables 11.1, 11.2, and 11.3).
In contrast, gray matter disorders manifest typically in early infancy with microcephaly, early severe seizures, and progressive dementia. Later features are axonal loss, retinal degeneration, progressive spasticity. The reflexes remain normal or become exaggerated. The EEG shows epileptiform discharges, the EMG and evoked potentials are usually normal. The main localization of grey matter loss is specific in some disorders. The basal ganglia are mainly involved in Juvenile Huntington Disease or Wilson Disease. Friedreich Ataxia involves spinocerebellar degeneration; in Spinal Muscular Atrophy, a genetic defect causes targeted death of neuronal cells in the anterior horn of the spinal cord with subsequent system-wide muscle wasting (see Tables 11.1, 11.2, and 11.3).
It is conceded that many neurodegenerative disorders have mixed white and gray matter involvement, or both grey and white matters are involved in a later stage of many of the disorders. Classification schemes may use the type of underlying molecular and genetic defect and change as such due to advances in identifying these defects. Examples are the group of Peroxisomal Disorders that include Adrenoleucodystrophy and Zellweger Syndrome, and the group of Lysosomal Disorders including Krabbe Leukodystrophy and Metachromatic Leukodystrophy).
Postnatal complications such as kernicterus, sepsis, meningitis, and head trauma should be investigated in the history as non-supportive of a neurodegenerative disorders. On the other hand, a positive family history of neurological disorders or early and unexplained deaths may suggest the presence of an undiagnosed inherited neurodegenerative disorder. Other confounders mimicking neurological regression and deterioration such as medical disorders, visual impairment, hearing loss, epilepsy, autism spectrum disorders, intellectual disability, attention-deficit hyperactivity disorders, or child abuse and neglect need to be considered when taking the history.
The clinical examination should include head circumference to detect megalencephaly, an important feature in Canavan, Tay-Sach’s, Sandhoff’s and Alexander Disease, or microcephaly that is a usual feature of many gray matter disorders due to progressive neuronal loss. Dysmorphic features need to be assessed as facial dysmorphisms are associated with some neurodegenerative disorders such as Zellweger Syndrome and mucopolysaccaridoses. A thorough neurological examination permits us to further define which specific nervous system functions or systems are deranged. In particular one should also assess ocular abnormalities that are prevalent in neurodegenerative disorders, in the form of optic atrophy in white matter disorders and retinal degeneration in grey matter disorders.
More specifically during the eye examination, look out for evidence of cortical blindness as in MELAS syndrome, abnormal movements (as in Ataxia telangiectasia, Gaucher’s disease, types 2 and 3, Kearns-Sayre syndrome and Pelizaeus-Merzbacher disease), corneal clouding (as in Wilson’s disease and Hurler’s disease), lens opacities (as in Fabry’s disease, Galactosemia, Lowe syndrome,) optic atrophy (as in Canavan disease Metachromatic leukodystrophy, Adrenoleukodystrophy and Pelizaeus-Merzbacher disease) or a Cherry Red Macula (as seen GM1 gangliosidosis, Tay-Sachs disease, Sialidosis type I).
Specific clinical findings give clues to the diagnosis. For example, hepatomegaly and/or splenomegaly are prominent in mucopolysaccaridosis, sphingolipidosis, peroxisomal and mitochondrial disorders. Progressive renal failure is found in Fabry Disease and Lowe Syndrome. Abnormalities of the hair (seen in Menkes syndrome, Biotinidase deficiency and mucopolysaccaridoses), the kidneys (as in Zellweger syndrome). Mucopolysaccaridosis, Friedreich Ataxia, and mitochondrial disorders are associated with cardiac disorder.
Investigations for Neurodegenerative Disorders
As a general rule, findings on history and physical examination should guide the selection of laboratory investigations as many neurodegenerative disorders have unique genetic, metabolic, or enzymatic markers. Facilities for a comprehensive workup for most neurodegenerative disorders are currently unavailable in Uganda with most assays having to be performed by overseas laboratories.
Initial laboratory studies should include blood analysis covering complete blood count, glucose, calcium, anion gap, electrolytes, renal function tests, ammonia, aminotransferases, lactic acid, pyruvic acid, uric acid and ketones. Urine analysis is done for ketones, pH, aminoaciduria, organic aciduria, homocystinuria, mycopolysaccharides and oligosaccharides. Serum ammonia, lactate, pyruvate, amino acids, and urine for amino acids and organic acids would screen for most amino acids disorders, organic acidopathies, and urea cycle abnormalities. Chest X-ray may show cardiomegaly in mitochondrial disorders, Friedreich Ataxia, and mucopolysaccharidosis. Conduction abnormalities may be present on electrocardiogram (ECG). Skeletal survey may reveal specific bony abnormalities such as dysostosis multiplex in mucopolysaccharidosis.
Children with dysmorphic features should have genotyping and chromosomal studies. Other investigations that bring important information are EEG often showing bilaterally synchronous paroxysmal discharges in grey matter disorders versus continuous non-paroxysmal slow wave activity in white matter disease. In diseases involving both the grey and white matter, the pattern may be mixed with both bilaterally synchronous paroxysmal discharges and markedly increased recordings of slow wave activity. The electroretinogram (ERG) may provide information about retinal involvement that features in several metabolic diseases. Recording of visual evoked potentials assist in documenting retinal lesions that may be more focal or restricted to retinal ganglion cells as in gangliosidosis. Brainstem auditory evoked potentials may be abnormal in a demyelinating disease or in axonal lesions. Nerve conduction studies show decreased nerve conduction velocity in demyelinating neuropathy, and decreased amplitude of the motor or sensory action potential in axonal neuropathy. Abnormal neurogenic changes can be differentiated from myopathic changes by analysis of the electromyogram.
Specific diagnostic tests and enzyme assays can be done to identify specific neurodegenerative disorders that often involve skin fibroblast culture, CSF examination, DNA studies, nerve, or muscle biopsies. These tests are not done routinely but only in specialized laboratory, are often expensive, and can be found in other literature [17].
Role of Neuroimaging
There are a range of neuroimaging techniques that could be employed for the early diagnosis of neurodegenerative disorders; however one needs to consider the purpose of the investigation. Early diagnosis may be used to appropriately delineate a specific disease condition from others that may present with similar clinical symptoms especially in the early stages, alternatively it may be utilized to timely identify a malfunctioning central nervous system before the clinical symptoms appear.
In Uganda there are limited diagnostic modalities with respect to brain imaging despite its significant value. Cranial computed tomography (cranial CT) and magnetic resonance imaging (MRI) brain scans are usually employed. MRI however plays a primarily vital role in diagnosis of central nervous system degenerative illnesses because it is exceptional in the visualization of many cerebral abnormalities compared to CT.
Other powerful neuroimaging techniques that permit visualization of organ structure and function with precision include: positron emission tomography (PET) and single photon emission computed tomography (SPECT) but these are currently unavailable. These utilize radio-ligands which measure in detail the functioning of distinct areas of the human brain and are beneficial in detecting and characterizing potential pathophysiological brain changes. These methods particularly PET that has the higher sensitivity are of great value in detecting early stages of the causes of cognitive impairment [18].
General Treatment Guidelines
Treatment can be directed towards the underlying disorder, associated features, and complications. Dietary restriction is useful in certain diseases such as phenylketonuria, maple syrup urine disease, adrenoleukodystrophy etc. Treatable complications include epilepsy, sleep disorder, behavioral problems, feeding difficulties, gastroesophageal reflux, spasticity, drooling, skeletal deformities, and recurrent chest infections. Specific treatments to counteract the offending metabolite (s), improve or decrease abnormal enzyme function, or to off-set metabolic dysfunction are possible for specific diseases in addition to organ transplantation including bone marrow transplantation (when irreparable brain damage has not occurred). Bone marrow transplantation has been shown variable but promising results in lysosomal storage diseases, mucopolysaccharidoses, Gaucher’s Disease, metachromatic leukodystrophy, and adrenoleukodystropy. It may be possible in the future to target specific pathways with somatic gene therapy but this will require more technological advances and discoveries. A referral for genetic counseling is important due to heritability of many neurodegenerative disorders. Public health initiatives should focus on the risk of consanguineous marriages as a way to raise awareness of a preventable cause of neurodegenerative disorders in childhood.
Common Etiologies of NDDD’s Seen in Uganda
Human Immunodeficiency Virus (HIV)
The human immunodeficiency virus (HIV) belongs to the Lentivirus genus of the Retroviridae family. The UN estimates that there were 35.3 (32.2–38.8) million people in the world infected with HIV in 2012 [19]. Uganda is estimated to have 1.5 million people living with HIV-1, of whom 190,000 are children [20].
HIV-1 is the most common cause of HIV infection in Africa with mother-to-child transmission of HIV (MTCT) being the major mode of acquisition of infection in children. The estimated risk of perinatal acquisition of untreated women to their infants ranges from 13 % to 30 % with approximately 400,000 new HIV-1 infections occurring each year [20].
Related posts:
 A Case Report of Mania and Glioma
Environmental Toxins as Causes of Brain Degeneration in Sub-Saharan Africa
A Case of Alzheimer’s Dementia in Uganda
Vitamin Deficiencies and Neuropsychiatric Disorders in Sub-Saharan Africa
A Case Report of Mania and Glioma
Environmental Toxins as Causes of Brain Degeneration in Sub-Saharan Africa
A Case of Alzheimer’s Dementia in Uganda
Vitamin Deficiencies and Neuropsychiatric Disorders in Sub-Saharan Africa
 Review of Reasons for Patients to Receive a CT of the Head and Neck Region in Uganda in 2011–2012
Review of Reasons for Patients to Receive a CT of the Head and Neck Region in Uganda in 2011–2012
 CT Findings in the Brain of Adult Patients with HIV/AIDS
CT Findings in the Brain of Adult Patients with HIV/AIDS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


