Fig. 15.1
MRI revealing a tuberculoma in the left pons area, in a patient with tuberculous meningitis, manifesting with paresis of n. abducens
15.3.3 Cerebrospinal Fluid
Detection of M. tuberculosis
The gold standard for diagnosis confirmation is demonstration of M. tuberculosis in the CSF by smear examination after Ziehl–Neelsen staining or culture. In CSF microscopy, acid-fast bacilli may be seen in up to 80 % of adult cases but only 15–20 % in children; large volumes (>6 ml) of CSF increase the diagnostic yield. Acid-fast bacilli are less frequently found in CSF with tuberculoma or spinal tuberculosis, compared to tuberculous meningitis (Marais et al. 2010; Thwaites et al. 2009).
Mycobacteria are slow-growing organisms; in conventional solid media (Lowenstein–Jensen), the growth can take up to 6–8 weeks. The BACTEC liquid media used in most microbiology labs has improved culture yields and enables detection of mycobacteria in average of 15 days. After anti-tuberculosis treatment is started, the sensitivity of smear or culture is lower. For susceptibility testing, direct culture methods are used, including liquid culture microscopic observation drug susceptibility (MODS), colorimetric redox indicator methods and the nitrate reductase assay (Thwaites et al. 2009; Garg et al. 2013).
Molecular-based PCR assay techniques including nucleic acid amplification tests (NAAT) are effective methods with high specificity but low sensitivity for rapid detection of mycobacterial DNA in CSF and other specimens. Molecular methods may be useful even in partially treated patients as mycobacterial DNA in CSF can be detected for 1 month after the initiation of the treatment (Solomons et al. 2014; Nhu et al. 2014; Garg et al. 2013). Quantitative RT-PCR for M. tuberculosis DNA and multiplex PCR using different primers have been introduced as possible useful methods for a rapid diagnosis (Takahashi et al. 2012; Kusum et al. 2011).
CSF Inflammatory Parameters
For the diagnosis of CNS tuberculosis, examination of CSF is mandatory; the volume of CSF is important, and repeated lumbar punctures may be needed. Typical CSF findings included in the consensus diagnostic criteria set are: a clear appearance, pleocytosis 10–500 cells/μL with a lymphocytic predominance of >50 %, elevated protein >1 g/L and decreased glucose levels (<2.2 mmol/L; CSF/plasma ratio <50 %), but atypical findings have been demonstrated. Lower protein levels and white cell counts or non-inflammatory CSF are more frequent in patients with HIV co-infection (Marais et al. 2010; Chamie et al. 2014; Cecchini et al. 2009).
Adenosine deaminase (ADA) is an enzyme, considered as an indicator of immunity, and a potential diagnostic marker to improve the diagnosis of tuberculous meningitis. In some studies, increased ADA levels in the CSF have been demonstrated in tuberculous meningitis but not in bacterial or viral meningitis; however, the results are inconsistent (Agarwal et al. 2014; Tuon et al. 2010).
15.4 Chronic Neuroborreliosis
15.4.1 Clinical Background
Lyme borreliosis, or Lyme disease, is a tick-borne illness caused by a spirochete Borrelia burgdorferi sensu lato complex that can result in dermatological, neurological, cardiac and musculoskeletal disorders. Lyme neuroborreliosis develops usually within 2–6 weeks after the tick bite, most frequently as lymphocytic meningoradiculitis (Bannwarth syndrome) in Europe, meningitis in the United States and facial nerve palsy in children. The number of cases of neuroborreliosis has increased in recent years that may be partially explained by higher incidence but also by improved diagnostic methods (Aguero-Rosenfeld et al. 2005; Stanek et al. 2012).
Chronic or late Lyme neuroborreliosis occurs in less than 5 % of neuroborreliosis patients, lasting for more than 6 months after tick-transmitted infection. Chronic neuroborreliosis is defined as a continuous active disease process with CNS inflammation and is characterised by various manifestations like recurrent or progressive encephalomyelitis with pareses, movement disorders, seizures, gait and bladder disturbances or stroke-like syndrome caused by vasculitis, radiculopathy or peripheral neuropathy (Ljostad and Mygland 2013; Hansen et al. 2013; Mygland et al. 2010). Psychiatric features include cognitive impairment, psychosis with visual and auditory hallucinations and obsessive–compulsive disorders. Depressive state among patients with late Lyme disease is reported to be common, ranging in 20–66 %. Whether the chronic Lyme neuroborreliosis is caused by the persistence of spirochetes in the brain leading directly to tissue damage or it is a result of immunological changes in the absence of continuing infection is still debated. It is believed that the pathological processes in neurosyphilis and neuroborreliosis are similar as spirochetes are known to be neurotrophic (Miklossy 2012). For treatment of chronic neuroborreliosis, intravenous penicillin G and ceftriaxone are empirically effective, and theoretically doxycycline could be as effective, but there is not enough evidence from trials. Response to antibiotic treatment in chronic Lyme disease is usually slow and may be incomplete (Ljostad and Mygland 2013; Hansen et al. 2013; Mygland et al. 2010).
15.4.2 Diagnosis of Chronic Neuroborreliosis
The European guidelines define chronic neuroborreliosis by the following criteria: (1) clinical syndrome with neurologic manifestation, without other possible causes; (2) CSF pleocytosis; and (3) intrathecally produced anti-Borrelia antibodies, except for peripheral neuropathy with antibodies in serum (Stanek et al. 2011, 2012; Mygland et al. 2010). A diagnosis of chronic Lyme disease based on persistence of non-specific symptoms without objective clinical and laboratory findings, which has been reported in the United States but also in other countries, cannot be supported (Ljostad and Mygland 2013; Feder et al. 2007; Cameron et al. 2004). Imaging is not essential for the diagnosis, but in chronic neuroborreliosis, MRI may reveal meningeal enhancement due to chronic inflammation or ischaemic-like white matter lesions (Agarwal and Sze 2009; Aalto et al. 2007).
15.4.3 Cerebrospinal Fluid
Detection of B. burgdorferi in CSF
Direct detection of the causing agent in later manifestation of Lyme borreliosis is limited. There is not enough evidence for microscope-based assays for use as a diagnostic tool, due to low sensitivity and specificity (Mygland et al. 2010). Recovery of B. burgdorferi sensu lato from CSF is complicated and can be done only in 10–17 % of untreated patients (Cerar et al. 2013; Aguero-Rosenfeld et al. 2005). Furthermore, by most conventional bacteriologic standards, borrelial cultures are poorly standardised, labour intensive, expensive and slow, requiring up to 12 weeks of incubation before being considered negative. Thus direct culture technique is hardly ever used in diagnosis of neuroborreliosis in clinical practice.
B. burgdorferi sensu lato DNA has been detected by PCR in CSF specimens, but it is of low diagnostic sensitivity, being positive in only 10–30 % cases with lower detection levels in longer duration of the disease that prevents implementation of this method in clinical practice (Aguero-Rosenfeld 2008; Wilske et al. 2007). The sensitivity of the PCR may depend also on the clinical presentations, CSF WBC count and whether antibiotic treatment is given or not. The spirochetes have been detected in the brain biopsy specimens and in autopsy samples (Miklossy 2012). PCR and CSF culture may be corroborative in the early stage of the disease when antibodies are absent or in immunosuppressed individuals, but otherwise not recommended for diagnosis (Mygland et al. 2010).
CSF Inflammatory Parameters
Most patients with ongoing neuroborreliosis regardless of clinical symptoms have increased WBC, typically 10–1,000 cells/mm3 (in 60 % in a range of 30 and 300 cells/mm3), mainly lymphocytes and plasma cells, with an exception in cases with peripheral neuropathies. Further supporting findings include elevated CSF protein up to 1–3 g/L or even higher and oligoclonal IgG bands. Unlike in early Lyme neuroborreliosis, chronic patients may have low CSF glucose levels. In contrast to other meningitides of bacterial origin, the CSF lactate concentration is in normal ranges (Ljostad and Mygland 2013; Hansen et al. 2013; Djukic et al. 2012; Stanek et al. 2011).
CSF Immunological Markers: Detection of Antibodies
Specific intrathecal production of IgM and IgG antibodies is one of the key features of neuroborreliosis (Ljostad and Mygland 2013; Stanek et al. 2012; Aguero-Rosenfeld 2008; Blanc et al. 2007). However, there are no uniformly accepted tests to measure Borrelia antibodies in CSF. Methods that have been used include capture immunoassays, CSF/serum antibodies determined by ELISA and Western immunoblots (Coyle et al. 1995, Wilske et al. 2007). Diagnostic yield of CSF antibodies could be improved when at least two antigens, flagella and one of the new antigens or two of the new antigens (decorin-binding protein A, BBK32, and outer surface protein C, and IR(6) peptide of borrelial VlsE protein) are used (Panelius et al. 2003). Positive intrathecal production of Borrelia-specific antibodies is indicated by an antibody index >1.3 (antibody index = CSF/serum Borrelia antibody concentration ratios: CSF/serum IgG concentration ratios, see also Chaps. 10 and 16); a pathological antibody index has a sensitivity of 80 % in early Lyme neuroborreliosis and nearly 100 % in longer duration (Wilske et al. 1986), Tumani et al. 1995). Intrathecal production can also be determined by testing CSF and serum at matching concentrations using immunoblotting. Of note anti-Borrelia antibodies may persist for years after the infection, and thus the specificity is low without other criteria of chronic neuroborreliosis (Djukic et al. 2012; Mygland et al. 2010; Wilske et al. 2007).
Detection of Chemokines and Cytokines in the CSF
Borreliae that enter the CNS are recognised by monocytes, macrophages or dendritic cells, which produce proinflammatory cytokines and induce chemokines. Increased concentrations of interleukin 4 (IL-4), IL-5, IL-6, IL-8, IL-10, IL-12, IL-18 and gamma interferon (IFN-γ) have been found in the CSF samples of patients suffering from Lyme neuroborreliosis (Cerar et al. 2013). Various chemokines (CXCL8, CXCL10, CXCL11, CXCL12, CXCL13) are released as a result of inflammation and stimulate chemotaxis involved in the CNS (Moniuszko et al. 2014). Studies have suggested that measuring CXCL13 enables to distinguish neuroborreliosis from other inflammatory diseases of the CNS and to evaluate treatment response (Cerar et al. 2013; Senel et al. 2010). However, it is not specific as described also in other viral and spirochetal infections such as tick-borne encephalitis (TBE), neurosyphilis and CNS lymphoma (Moniuszko et al. 2014; Marra et al. 2010; Schmidt et al. 2011). The exact diagnostic cut-off values, however, are not established, and thus potential diagnostic usefulness of CXCL13 levels in CSF in chronic neuroborreliosis is still debatable (Ljostad and Mygland 2013; Bremell et al. 2013; Schmidt et al. 2011).
15.5 Tick-Borne Encephalitis (Chronic Form)
15.5.1 Clinical Background
Tick-borne encephalitis (TBE) is caused by the tick-borne encephalitis virus (TBEV), which is a single-stranded RNA virus that has European, Siberian and Far-Eastern subtypes. The course of the European subtype is mainly asymptomatic or if symptomatic then characterised by a biphasic course with development of meningitis, meningoencephalitis or myelitis which can be followed by a postencephalitic syndrome. The case fatality rate in the European subtype is low around 1–2 % (Holzmann 2003; Dumpis et al. 1999). In contrast, in the Far-Eastern subtype, the disease is mostly monophasic but causes more severe forms with neurological sequelae and the fatality rates of up to 30 %. The Siberian subtype characteristically induces a less severe acute period with a case fatality rate of 6–8 %, but there is a tendency to develop chronic TBE (Dumpis et al. 1999).
Acute forms of tick-borne encephalitis (TBE) are described in Chap. 14.a.i. A chronic form of TBE mainly affects people <35 years and has been associated exclusively with the Siberian subtype of TBEV in Siberia and Far East. Hyperkinesias (54 %), epilepsia partialis continua, encephalopoliomyelitis and lateral amyotrophic sclerosis syndrome have been described (Mukhin et al. 2012; Poponnikova 2006; Gritsun et al. 2003a; Nadezhdina 2001). In the chronic form of TBE, a relapsing course following the acute TBE has been described or delayed development of the progressive disease after a long incubation period without the typical acute stage (Gritsun et al. 2003b; Frolova et al. 1987; Vasilenko and Grigor’eva 1987). Chronic TBE is very rare in Europe, with only two cases reported in Lithuania (Mickiene et al. 2002). Experimental studies have demonstrated the ability of TBEV Siberian subtype to persist and produce the chronic disease in animal models (Gritsun et al. 2003a; Frolova et al. 1987). The chronic form of TBE has been associated with mutations in the TBEV NS1 gene (Gritsun et al. 2003b) and immunological changes with defective T-cell response and imbalance in production of cytokines (Naslednikova et al. 2005).
The diagnostic criteria of chronic TBE are poorly defined, and there is no uniform consensus.
15.5.2 Cerebrospinal Fluid
Detection of TBEV in CSF
TBEV is very rarely if ever isolated from CSF. The RT-PCR to detect TBE RNA in CSF has not been successful either (Holzmann 2003). In a study in which TBE RNA was successfully detected in all blood samples, only 1 of 31 CSF samples tested positive during early viraemic period (Saksida et al. 2005). In fatal cases, the virus has been detected by RT-PCR in the brain tissue (Holzmann 2003). A strain of the Siberian subtype of TBEV was isolated postmortem from a patient who died of a progressive (2-year) form of TBE 10 years after being bitten by a tick, and another strain was detected in CSF of a 11-year-old girl with relapsing course of epilepsia partialis continua during 5 years (Gritsun et al. 2003a).
CSF Inflammatory Parameters
Patients with TBE have shown CSF pleocytosis in the range of 12–1,100 cells with a predominance of segmented granulocytes (60–70 %) over lymphocytes (30–40 %) and a moderate impairment of the blood–CSF barrier (increased CSF–to–serum albumin ratio) (Holzmann 2003).
Detection of Antibodies in CSF
The diagnosis of TBE is based on the demonstration of TBE-specific IgM and IgG antibodies in blood, which are usually detectable at the beginning of the second phase and increase to high titres. IgG antibodies persist over a lifetime and prevent reinfection. In CSF, intrathecally produced specific antibodies can only be found in 50 % of patients early after the disease onset, but within 10 days, they almost invariably become detectable (Holzmann 2003). However, there are limited data on antibodies in chronic TBE. In two patients in Siberia with progressive or relapsing course of chronic TBE, the TBEV Siberian serotype strains were isolated, but virus-specific antibodies were not detected that might show a possible defect of immune mechanisms, contributing to the persistence of the virus (Gritsun et al. 2003b).
15.6 Prion Diseases
15.6.1 Clinical Background
Prion diseases are rare fatal transmissible neurodegenerative disorders, characterised by the accumulation of protease-resistant prion protein (PrP), an isoform of a normal cellular protein. Aetiologically, human prion diseases can be divided into sporadic, inherited and acquired forms, with a diversity of clinical presentations (Araujo 2013; Wadsworth and Collinge 2007).
About 85 % of prion disease cases occur as sporadic Creutzfeldt–Jakob disease (CJD), with a yearly incidence rate of 1–2 cases per million of population. Clinically, it presents with rapidly progressive dementia, fast development of akinetic mutism, along with generalised myoclonus, ataxia, extrapyramidal and pyramidal signs and cortical blindness. Inherited cases that are classified as familial CJD, Gerstmann–Sträussler–Scheinker syndrome or fatal familial insomnia are associated with mutations in the human prion protein gene. Iatrogenic CJD may occur due to transmission through medical procedures. Variant CJD represents bovine-to-human transmission associated with the consumption of meat products that was described first in 1996 in the United Kingdom. Around 230 variant CJD cases have been reported, and global monitoring continues through the EuroCJD network. Kuru was endemic in Papua New Guinea, transmitted by ritual cannibalism and manifested after a long incubation period with ataxia and dementia developing later (ECDC 2013; Araujo 2013; Wadsworth and Collinge 2007).
Human prion diseases are ultimately fatal – death follows symptom onset within 2 years. The therapeutic approaches include polyanionic and polycyclic drugs to prevent conversion of PrP and laminin receptor antagonists that have provided some experimental promises. Active and passive immunisation against PrP in animals has shown divergent effects and can trigger neurotoxic pathways (Panegyres and Armari 2013; Aguzzi et al. 2013).
15.6.2 Diagnosis of Creutzfeldt–Jakob Disease
Diagnostic criteria for CJD were formulated in 1979 and updated in 2009, including clinical criteria and tests/investigations. For probable or possible sporadic CJD, two clinical signs of the following four should be fulfilled: (1) dementia, (2) cerebellar or visual signs, (3) pyramidal or extrapyramidal signs and (4) akinetic mutism. Additionally, for a probable case, one of the following tests should be positive: (1) periodic sharp wave complexes in EEG that are of high specificity and low sensitivity or (2) 14-3-3 protein in CSF that has a high sensitivity but modest specificity or (3) high signal abnormalities in the caudate nucleus and putamen or at least two cortical regions in MRI (Fig. 15.2). However, the accuracy of diagnostic criteria has been assessed in a series of rapid progressive dementia and the level of the tau protein considered as a possible additional marker for the diagnosis (Felix-Morais et al. 2014; Tagliapietra et al. 2013; Zerr et al. 2009).
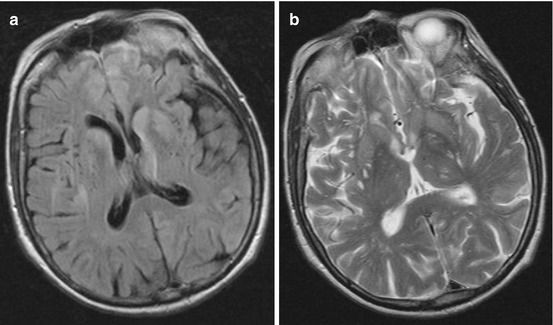
Fig. 15.2
MRI in sporadic CJD demonstrating bilateral hyperintensities in striatum and frontomedial cortex: (a) FLAIR; (b) T2
For variant CJD, the case definition for reporting communicable diseases has been approved in the Commission Implementing Decision of the European Council in 2012, combining preconditions of medical history, of at least four clinical criteria out of five (early psychiatric symptoms; painful sensory symptoms; ataxia; myoclonus or chorea or dystonia; dementia), epidemiological criteria and negative EEG findings without periodical complexes typical for sporadic CJD. The diagnostic criteria for a confirmed case are neuropathological findings: spongiform degeneration, neuronal loss, astrocytosis and PrP immunohistochemistry (EC 2012).
15.6.3 Cerebrospinal Fluid
Detection of Prions
PrP cannot be detected in CSF by currently available methods. A promising novel method of real-time quaking-induced conversion has been developed for detection of PrP in the diluted CJD brain homogenate, but further studies are needed for more evidence (Atarashi et al. 2011).
CSF Inflammatory Parameters
Typically there are no changes in inflammatory parameters in CSF in prion diseases, but a routine CSF examination may be of importance for differential diagnosis to exclude potentially treatable inflammatory conditions. A large study on standard CSF examination showed isolated mild increases of white cell count or total protein in a small proportion of patients with sporadic CJD. The presence of >20 cells/μL or 1 g/L of protein in CSF suggests an alternative diagnosis (Green et al. 2007).
CSF Immunological Markers
In patients with rapidly progressive dementia and suspected sporadic CJD, it is recommended to examine CSF to detect 14-3-3 protein as a marker with a high sensitivity of 92 % and specificity of 80 % for CJD (Muayqil et al. 2012; Stoeck et al. 2012). However, the positive results have also been reported in other dementias and encephalopathies, infections and cerebrovascular diseases and metastases (Deisenhammer et al. 2009). Other brain-derived proteins have also been considered as diagnostic markers for CJD. In a Swedish study, the combination of t-tau levels and t-tau to p-tau ratios in CJD patients had a high specificity against important differential diagnoses (Skillback et al. 2014). However, CSF 14-3-3 protein had higher sensitivity than tau protein and S100b in the United Kingdom 10-year review (Chohan et al. 2010).
15.7 Progressive Multifocal Leucoencephalopathy
15.7.1 Clinical Background and Diagnosis
Progressive multifocal leucoencephalopathy (PML) is a potentially fatal CNS infection with progressive deterioration, caused by a polyoma virus (JC virus/John Cunningham virus). Polyoma viruses are double-stranded enveloped DNA viruses. Subclinical infection may occur in childhood, and about a half of an adult population with sclerosis multiplex have been tested as seropositive for JCV (Steiner and Berger 2012). PML occurs as an opportunistic infection in patients with monoclonal antibody immunosuppressive therapies, HIV infection or lymphoproliferative disorders. It has been reported in association with natalizumab treatment of multiple sclerosis, rituximab for lymphoproliferative and rheumatic disorders, efalizumab for psoriasis and brentuximab vedotin therapy (Carson et al. 2014; Mancuso et al. 2012; Mentzer et al. 2012; Carson et al. 2009). There is no antiviral therapy against the JCV, and the prognosis is poor. Highly active antiretroviral therapy has a beneficial impact to the prognosis of PML in HIV patients (Casado et al. 2014; Steiner and Berger 2012).
The proposed case definition is based on detection of JCV DNA in CSF or positive DNA and viral antigens with typical histopathology in the brain biopsy or autopsy tissue, in appropriate clinical settings and in brain MRI findings. Clinically, cognitive and behavioural disorders, pareses, ataxia, sensory loss, speech and visual disturbances and seizures have been described. PML damage is related to the brain demyelination, and MRI typically reveals lesions in subcortical and periventricular white matter (Fig. 15.3), cerebellum or peduncles, but also grey matter involvement can occur rarely. Typical histopathological findings include enlarged oligodendroglial nuclei, bizarre astrocytes and demyelination (Berger et al. 2013; Mentzer et al. 2012; Steiner and Berger 2012).
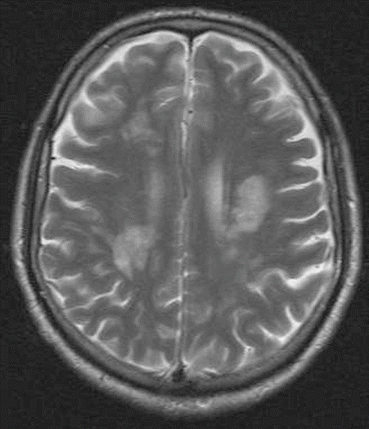
Fig. 15.3
MRI in a PML patient showing lesions in periventricular and deep white matter
The 2-step enzyme-linked immunosorbent assay (ELISA) assay is used to detect the anti-JCV serostatus in patients with multiple sclerosis, as a tool for PML risk stratification, but not for confirmation of the diagnosis of PML (Gorelik et al. 2010).
15.7.2 Cerebrospinal Fluid
In a routine CSF examination, most PML patients demonstrate a normal cell count (usually less than 20 cells/μL) but mildly increased protein levels (Berger et al. 2013). The JCV DNA can be detected by PCR in a laboratory with specific expertise. However, positive PCR without typical clinical and radiological findings is not sufficient to provide evidence for the diagnosis, and a negative JCV PCR does not exclude PML. In HIV-infected patients who have developed PML-immune reconstitution inflammatory syndrome as a result of antiretroviral therapy, detection of JCV declines substantially. Repeated testing of CSF may be needed, but testing of blood or urine for JCV has no diagnostic value (Berger et al. 2013; Mentzer et al. 2012; Fong et al. 1995). A novel method for mutation scanning has been developed with the potential to serve as an additional diagnostic method when combined with routine RT-PCR testing (Nakamichi et al. 2014).
A CSF JCV antibody index has been suggested as a complementary tool for the diagnosis of natalizumab-associated PML in cases with low levels of JC virus DNA in CSF, but it needs additional evidence (Warnke et al. 2014).
15.8 Subacute Sclerosing Panencephalitis
15.8.1 Clinical Background and Diagnosis
Subacute sclerosing panencephalitis (SSPE) is a chronic encephalitis secondary to measles infection that causes demyelination damage of CNS. The measles virus (a single-stranded, negative-sense, enveloped RNA virus in the family of Paramyxoviridae) occurs predominantly in regions with low vaccination rates. The SSPE clinical manifestations occur on average 6 years after measles and include cognitive and behavioural disorders, myoclonic seizures, pareses, rigidity and akinetic mutism, rarely cerebellar signs and speech disorder and, in some patients, early visual symptoms with macular and retinal changes. SSPE is more prevalent in males, and the presentations usually occur in childhood, but adult cases have been described as well. The incidence of SSPE is decreasing as a result of measles vaccination (Colpak et al. 2012; Erturk et al. 2011; Gutierrez et al. 2010).
The diagnosis of SSPE is based on clinical manifestations, EEG findings with periodic complexes, demyelinating lesions in MRI and immunological evidence of intrathecal anti-measles antibody production. Clinically, measles have been demonstrated by a history in about half of SSPE patients (Gutierrez et al. 2010; Lakshmi et al. 1993).
There is no effective treatment for SSPE. Oral isoprinosine and intrathecal alpha-interferon may prolong survival but do not change the long-term outcome. The prognosis of SSPE is poor, with a lethal outcome in a majority of cases. The best method for prevention of SSPE is vaccination against measles (Gutierrez et al. 2010; Eroglu et al. 2008).
15.8.2 Cerebrospinal Fluid
In CSF examination the SSPE patients have a mild pleocytosis, normal or elevated protein and normal glucose CSF-to-serum ratio. Diagnosis of SSPE is confirmed by CSF anti-measles antibodies; ELISA for CSF antibodies has a sensitivity of 100 % and specificity of 93 % and a positive predictive value in appropriate clinical settings (Gutierrez et al. 2010; Lakshmi et al. 1993). IgG titre range in CSF has been demonstrated from 1:40 to 1:1,280 and the CSF-serum IgG ratio from 5:1 to 40:1 (Manayani et al. 2002). Results of studies on CSF cytokine levels show discrepancies between reports (Aydin et al. 2010).
15.9 Toxoplasmosis
15.9.1 Clinical Background
Toxoplasmosis is caused by the obligate intracellular protozoal parasite Toxoplasma gondii, transmitted by the oral or transplacental route. Common sources of human infection are consumption of undercooked lamb or pork meat that contains viable tissue cysts or direct ingestion of oocytes from contaminated soil, water, goat’s milk or unwashed vegetables (Finsterer and Auer 2013). In immunocompetent adults, most T. gondii infections are subclinical, but severe clinical manifestations occur in immunocompromised patients (Patil et al. 2011). The prevalence rates of latent Toxoplasma infections in HIV-infected patients have been found to vary greatly from 3 to 97 % (Ammassari et al. 2000). Prior to the introduction of effective antiretroviral therapy, about half of seropositive patients had cerebral toxoplasmosis (Oksenhendler et al. 1994).
The involvement of cerebral grey or white matter in these patients results in encephalitis or a CNS mass lesion, which manifests clinically as headache, epilepsy, hemiparesis, psychosis, cognitive dysfunction or movement disorders. The basal ganglia, thalamus and corticomedullar junction are most frequently affected (Finsterer and Auer 2013). Neurotoxoplasmosis is diagnosed upon clinical, serological, CSF or imaging investigations. MRI findings include lesions with mass effect in cerebral cortex, subcortical white matter, basal ganglia, brain stem or cerebellum (Fig. 15.4).
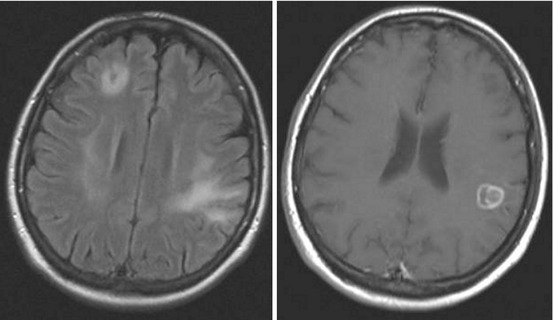
Fig. 15.4
MRI of a HIV patient with toxoplasmosis, revealing lesions in corticomedullar junction
15.9.2 Cerebrospinal Fluid
The combination of mononuclear pleocytosis, elevated protein count and normal glucose levels in CSF of immunocompromised patients is highly suggestive of toxoplasma encephalitis.
Detection of T. gondii in CSF
A definite diagnosis of cerebral toxoplasmosis could be made by histologic demonstration of tachyzoites in brain biopsies taken from lesions. This procedure is hardly ever used in clinics due to the high rate of serious complications. Furthermore the sensitivity of tissue cultures that have been used in the past was lower than of PCR (Dupon et al. 1995). However, the PCR for T. gondii DNA in CSF has shown variable sensitivity values from 0 to 100 % that depend on various factors, including the site of a puncture: studies have shown that the sensitivity of samples taken by ventricular tap is higher than those taken by lumbar puncture (Adurthi et al. 2011; Contini 2008; Contini et al. 1998). Direct identification of T gondii tachyzoites in CSF of adult patients is apparently extremely rare. In all but one reported case, the organisms have been identified in the ventricular rather than lumbar specimen (Brogi and Cibas 2000; Palm et al. 2008).
Detection of Antibodies in CSF
It is believed that more than 50 % of patients with cerebral toxoplasmosis present specific antibodies in the CSF due to permeability changes of the blood–CSF barrier allowing passive passage of antibodies from serum to the CSF (Dannemann et al. 1992). CSF serology appears to be remarkably sensitive for the diagnosis of cerebral toxoplasmosis (Adurthi et al. 2011) but has relatively low specificity in discriminating between recent, active and past dormant toxoplasma infection (Luft and Remington 1988). This could be overcome by measuring antibodies against excretory–secretory antigens which are the majority of the circulating antigens in sera in patients with acute toxoplasmosis. These antibodies measured in CSF by ELISA or immunoblotting appear to be good markers for cerebral toxoplasmosis also in patients with HIV infections (Meira et al. 2011).
15.10 Sleeping Sickness/African Trypanosomiasis
15.10.1 Clinical Background and Diagnosis
African trypanosomiasis is a parasitosis, caused by the protozoa Trypanosoma brucei gambiense (West African form) or Trypanosoma brucei rhodesiense (East African form), transmitted by tsetse flies. Incidence of African trypanosomiasis has decreased, but the WHO estimates a large population being at risk, and has classified this as a neglected tropical disease by 2020 (WHO 2014b; Truc et al. 2012). Sleeping sickness is endemic to Sub-Saharan Africa with the highest number of cases in Congo, but an increasing number of cases has been reported in non-endemic areas including Europe and North America, in travellers, military personnel and immigrants (Migchelsen et al. 2011).
There are two clinical stages of African trypanosomiasis – the first or haemolymphatic stage starts with fever, lymphadenopathy and chancres, followed by invasion of protozoa into CNS. The second or meningoencephalitic stage is characterised by severe headaches, disruption of the circadian rhythm with daytime somnolence and night time insomnia, cognitive and behavioural disorders, pareses and movement disorders. The disease eventually may progress to apallic syndrome, coma and death (Migchelsen et al. 2011; Brun et al. 2010).
For laboratory large-scale screening of the populations at risk, the card agglutination test for trypanosomiasis/T. b. gambiense (CATT) in serum is used, and the diagnosis is confirmed by microscopic verification of the parasite, followed by CSF examination for staging of the disease (WHO 2014b). Novel rapid serological tests have been introduced recently, but they need further evaluation of their diagnostic accuracy (Buscher et al. 2013). MRI may show diffuse white matter abnormalities, basal ganglia hyperintensities and ventricular enlargement, but neuroimaging is often not available in endemic areas (Brun et al. 2010; Kennedy 2008).
African trypanosomiasis is fatal if untreated, but current anti-trypanosomal therapies have limitations due to toxicity. Pentamidine or suramin are recommended for the first stage and melarsoprol and eflornithine for the neurological stage of sleeping sickness (WHO 2014b; Migchelsen et al. 2011; Kennedy 2008).
15.10.2 Cerebrospinal Fluid
Examination of CSF is essential for the diagnosis of human African trypanosomiasis with CNS involvement and also for selection of treatment and post-treatment follow-up. A pleocytosis >5 cells/μL is an indicator of CNS infection, but typically there are 100–300 cells/μL of lymphocytic origin. A slightly elevated cell count may persist for several months even after successful treatment. The microscopic detection of trypanosomes in CSF is diagnostic for the meningoencephalitis stage of the disease. Molecular amplification tests to detect trypanosomal DNA are sensitive; however, the accuracy of these tests as diagnostic tools has not been fully verified (Mugasa et al. 2012). Increased IgM concentrations in CSF are an early and specific diagnostic marker, and the intrathecal IgM response is higher than the IgG response, but detection of trypanosome specific antibodies is of low sensitivity (Brun et al. 2010; Lejon and Buscher 2005). Novel promising biomarkers including neopterin have been proposed for stage determination of human African trypanosomiasis but need further evidence (Tiberti et al. 2013; Burchmore 2012). CSF cytokines have not demonstrated sufficient sensitivity to be of clinical value for staging of the disease (MacLean et al. 2012).
15.11 Nipah Encephalitis
The Nipah virus along with the Hendra virus are the members of a newly identified genus of emerging paramyxoviruses, henipaviruses. Since their discovery in the 1990s, henipavirus outbreaks have been described mainly in Asian countries. The viruses have high economic and public health threat potential. When compared to other paramyxoviruses, henipaviruses appear to have unique characteristics. They are zoonotic viruses with a broader tropism and host range than most other paramyxoviruses and can cause severe acute encephalitis with unique features among viral encephalitides. Both henipaviruses are very likely to be transmitted to their amplifying or dead-end host (pigs, horses) by several species of fruit bats (Vigant and Lee 2011). The Nipah virus infects a wide range of mammalian species (pigs, dogs, goats and cats). It is extremely contagious among pigs and is readily transmitted to humans (Parashar et al. 2000). Moreover, direct transmission to humans and between humans has been reported (Chua 2012). Human may also become infected through consumption of raw fruit or date palm juice contaminated with the virus.
One of the most unusual features of Nipah infection is the propensity for patients to develop relapsing or late-onset encephalitis up to 53 months after acute infection (median 8 months); some patients may have a second relapse. Late-onset encephalitis, in patients who do not initially have neurological symptoms, occurs in about 3 % of cases. Eight percent of survivors of acute encephalitis have recurrent neurological disease (relapsed encephalitis). Patients with relapses are likely to have fever (46 %) and headache (42 %) and more likely to have seizures (50 %) and focal neurological signs (42 %) than those with acute encephalitis (Tan and Chua 2008). As in acute encephalitis, CSF is usually abnormal with mild lymphocytic pleocytosis. However, the absence of the virus in CSF suggests that the pathophysiology of acute and relapsed or late-onset encephalitis is different but needs further investigation. MRI findings usually involve damage of grey matter. As many as 35–75 % of symptomatic Nipah virus infections are fatal, and approximately 15 % of survivors have neurological sequelae (Sawatsky et al. 2007).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


