, Keiichi Niikura2, Akira Yamashita2, Daigo Ikegami2, Naoko Kuzumaki2, Michiko Narita2 and Tsutomu Suzuki3
(1)
Life Science Tokyo Advanced Research Center (L-StaR), 2-4-41 Ebara, Shinagawa-ku, Tokyo 142-8501, Japan
(2)
Department of Pharmacology, Hoshi University School of Pharmacy and Pharmaceutical Sciences, 2-4-41 Ebara, Shinagawa-ku, Tokyo 142-8501, Japan
(3)
Department of Toxicology, Hoshi University School of Pharmacy and Pharmaceutical Sciences, 2-4-41 Ebara, Shinagawa-ku, Tokyo 142-8501, Japan
Abstract
μ-Opioid agonists (μ-agonists), including morphine, are often used to treat the pain associated with cancer moderate-to-severe pain and moderate-to-severe noncancer pain due to other causes. However, μ-agonists also have various side-effects, in addition to potential for abuse or addiction. Recent clinical studies have shown that when μ-agonist analgesics are used appropriately to control pain, abuse and addiction usually do not develop. This chapter highlights recent findings regarding molecular adaptations observed in models of sustained pain, and discusses how such adaptations could reduce the abuse potential of μ-agonists under chronic pain.
Introduction
μ-Opioid agonists are the main analgesics used to treat the pain associated with cancer pain and moderate-to-severe noncancer pain due to other causes [1–3], even though they may be the subject of abuse and/or addiction. Due to this potential, their use for the treatment of severe pain has become complicated. However, it has been reported that when high levels of somatic pain are treated with μ-agonists, abuse and addiction rarely develop [1, 4, 5].
This review presents recent experimental findings on the molecular and neurobiological changes that result from exposure to chronic pain stimuli [6–11]. These findings suggest that, due to these changes, the abuse potential of μ-agonists is reduced when they are used as analgesics under clinical pain conditions.
Usefulness of μ-Opioids for the Treatment of Chronic Pain
Most clinically important opioid analgesics target μ-opioid receptors. μ-Opioid receptors have been shown to transduce signals through pertussis toxin (PTX)-sensitive Gi/Go proteins to inhibit adenylate cyclase, increase membrane K+ conductance, reduce Ca2+ current [12], and activate a phospholipase C (PLC)-IP3 pathway depending on the stimulation of βγ subunits [13–15].
The typical μ-opioid receptor agonist morphine is considered a “gold standard” by the World Health Organization (WHO) for the treatment of moderate-to-severe cancer pain. In 1965, fentanyl, an anilidopiperidine-class μ-opioid agonist, was reported as a potent synthetic analgesic. Fentanyl has a high affinity for μ-opioid receptors and its analgesic activity is 50–100 times greater than that of morphine. Since fentanyl has a low molecular weight and is lipid-soluble, it can be delivered transdermally. On the other hand, oxycodone, which has been in clinical use for many years, is a semisynthetic μ-opioid analgesic that was obtained from the naturally occurring alkaloid thebaine. Oxycodone has been shown to have a structure and lipid-solubility comparable to those of morphine and is comparable or only slightly inferior to morphine with regard to its analgesic potency [16]. In the clinic, the peripheral administration of oxycodone is uniquely effective for relieving some symptoms of pain in patients with neuropathic pain. Thus, μ-opioids are considered to be the drug of choice for the treatment of patients with chronic pain.
Role of the Mesolimbic Dopamine System in Opioid Reward
μ-Opioid receptor agonists produce euphoria in human subjects and act as positive reinforcers in several species, i.e., they induce drug-seeking behavior, which promotes their administration. Indeed, their reinforcing effects may become so strong that they become the primary motivation for behavior, which can lead to compulsive drug-seeking or addiction [17–19].
Many studies have suggested that this effect originates in the mesolimbic dopaminergic system. The mesolimbic dopaminergic system originates in the ventral tegmental area (VTA) of the midbrain and projects to the nucleus accumbens (NAc.), which is closely associated with the limbic system. It plays a role in the rewarding effects of intracranial self-stimulation, in the actions of abused drugs, including opioids, and in the intake of natural rewards such as food and water [20, 21]. μ-Opioid receptors in the VTA are known to be critical for the rewarding effects of opioids. For example, in an electrophysiological study, the systemic administration of morphine increased the firing rate of dopaminergic neurons in the VTA [22]. Furthermore, μ-opioids have been shown to increase both the release of dopamine and dopamine metabolites in the mesolimbic dopamine terminal fields [23–27].
The conditioned place preference (CPP) paradigm is an experimental paradigm that has been widely used to investigate the conditioned reward effects of drugs (e.g., μ-agonists) by repeatedly pairing them with a novel environment. After such repeated pairing, an experimental animal will spend more time in the environment that was paired with a rewarding drug than in a complementary environment that was paired with the vehicle. This is considered a drug-induced place preference. Use of the CPP paradigm has shown that intra-VTA injection of morphine produces a rewarding effect, and this effect is blocked by systemic administration of naloxone [28] or intra-VTA administration of naloxone methiodide [29]. This morphine-induced place preference can also be inhibited by the administration of dopamine antagonists or by neurochemical destruction of the NAc. [30, 31]. Furthermore, intra-VTA administration of the μ-opioid receptor antagonist D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP) or naloxone can induce place aversion, and these effects are inhibited by the intra-NAcc. Injection of 6-hydroxydopamine to induce the lesion of dopamine neurons [17]. Thus, dopamine-containing neurons in the midbrain VTA, which contains a high density of μ-opioid receptors, play important roles in the rewarding and aversive effects of μ-opioids.
Absence of Dependence-Liability of Opioids Under Chronic Pain
Opioid addiction is often not observed when opioids are used to treat pain. In some patients, withdrawal signs are only rarely observed when the opioid drug is withdrawn gradually after their pain is relieved. This observation supports the notion that opioids can be safely used to treat severe acute, cancer, and chronic pain [32, 33]. Studies in humans and animals support the idea that pain attenuates opioid-associated reward and euphoria [6, 7, 9, 11, 34, 35].
Chronic Pain Leads to Dysfunction of the Mesolimbic μ-Opioidergic System
In our experimental findings, spontaneous activity of the mesolimbic dopaminergic system and the release of dopamine in the NAc. after either the electrical stimulation of VTA neurons or systemic treatment with morphine are markedly suppressed by sciatic nerve ligation [9, 35, 36] (see Fig. 4.1), an animal model of neuropathic pain, which is caused by the persistent application of noxious stimuli, such as heat, cold, or chemicals, or acute nerve injury. Under these conditions, we also measured the changes in the ability of morphine to activate G proteins in the limbic forebrain, which contains the NAc., the lower midbrain, which contains the VTA, and the pons and medulla regions of both sham-operated and sciatic nerve-ligated mice by monitoring the binding of [35S]GTPγS to membranes [35]. Morphine concentration dependently increased the binding of [35S]GTPγS to membranes in the mouse limbic forebrain, lower midbrain including the VTA, and pons/medulla through μ-opioid receptors. Interestingly, the morphine-induced increase in the binding of [35S]GTPγS to membranes in the lower midbrain, but not the limbic forebrain or pons/medulla, was dramatically reduced by ligation of the sciatic nerve [35]. However, there was no difference between sham-operated and sciatic nerve-ligated mice with regard to the production of μ-opioid receptor in the lower midbrain [35]. This suggests that the neuropathic pain that is caused by ligation of the sciatic nerve ultimately reduces μ-opioid receptor function in the VTA. This selective reduction in μ-opioid signaling in the VTA is compatible with a reduction in the rewarding effects of μ-agonists, with the relative maintenance of analgesic effects, under neuropathic pain (see Fig. 4.1).
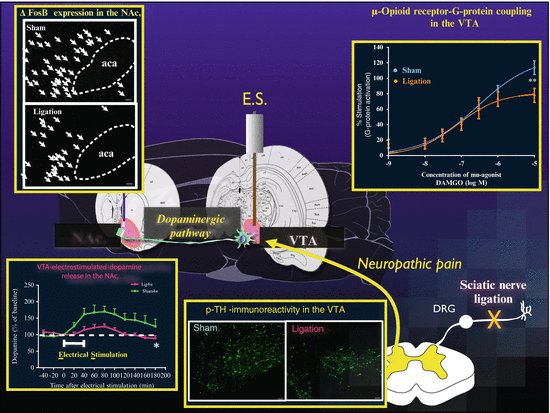
Fig. 4.1
Changes in the phasic activity of the mesolimbic dopamine system under neuropathic pain. μ-Opioid receptor-G-protein coupling in the VTA: Concentration–response curve for the effect of DAMGO on the binding of [35S]GTPγS to membranes of the lower midbrain from mice after sciatic nerve ligation or sham operation. p-TH immunoreactivity in the VTA: Immunofluorescent staining for phosphorylated-tyrosine hydroxylase (Ser31) (p-TH)-like immunoreactivity (IR) in the VTA of sham-operated or nerve-ligated rats. The images show p-TH-like IR in the VTA of nerve-ligated (Right) or sham-operated rats (Left). VTA-electrostimulated-dopamine release in the NAc.: Change in the dialysate levels of dopamine induced by electrical stimulation (ES) in the VTA. Effect of ES on dialysate dopamine levels in the NAc. ΔFosB expression in the NAc.: Images of ΔfosB-immunoreactivities in the NAc. of sham-operated or nerve-ligated animals. Scale bars = 50 μm. Figure adapted from [36], the author’s original work
One possible explanation for the decrease in μ-opioid receptor signaling in the VTA under chronic pain is a sustained increase in the release of β-endorphin, an endogenous μ-opioid. Prolonged exposure to β-endorphin could result in the phosphorylation of μ-opioid receptors, the uncoupling of these receptors from effector systems, and ultimately desensitization. Notably, β-endorphin has a greater desensitizing effect than morphine [37]. G protein receptor kinase 2 (GRK2), a serine/threonine kinase, promotes the phosphorylation induced by μ-agonists [38]. In one study, nerve-ligated mice showed an increase in the level of membrane-bound GRK2 in the VTA, but not the pons or medulla [35]. This increased level of GRK2 in the VTA might suppress μ-opioid receptor function under ligation of the sciatic nerve associated with the increased release of β-endorphin [35].
The Change in the Release of β-Endorphin Under Chronic Pain Is Critical for Downregulation of the Mesolimbic μ-Opioidergic System
As alluded to above, in the presence of pain, β-endorphin is released in various regions of the brain, including the mesolimbic pathway [39, 40]. Interestingly, the intra-VTA administration of a specific antibody to β-endorphin counteracts the suppression of the place preference produced by the intra-VTA administration of DAMGO (a μ-selective agonist) under neuropathic pain [43]. Furthermore, neither the finding that sciatic nerve ligation suppresses the place preference induced by the systemic administration of morphine nor the parallel decrease in the DAMGO-stimulated binding of [35S]GTPγS in the VTA are observed in β-endorphin knockout mice [36]. Furthermore, the finding that ligation of the sciatic nerve inhibits the morphine-induced release of dopamine in the NAc. is also not observed in β-endorphin knockout mice [36] (see Fig. 4.2). These findings suggest that the selective and sustained activation of mesolimbic β-endorphin might be important for suppressing the rewarding effects of exogenous μ-agonists under chronic pain.
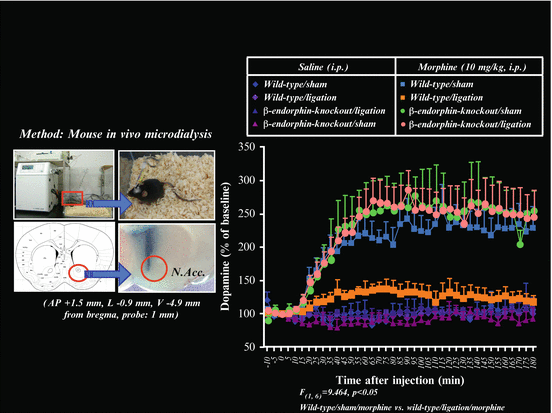
Fig. 4.2
Change in the increased dialysate dopamine level induced by morphine in β-endorphin knockout (−/−) mice. Localization of microdialysis probe sites in the mouse NAcc. (Left). Effects of treatment with morphine on the dialysate dopamine level in the NAcc. in sham- or nerve-ligated wild-type mice and β-endorphin knockout (−/−) mice (Right). Morphine (10 mg/kg, s.c.) or saline was injected at time 0. The data are expressed as percentages of the corresponding baseline levels with S.E.M. p < 0.001; sham-operated wild-type mice treated with morphine vs. sham-operated β-endorphin knockout (−/−) mice treated with morphine. Figure adapted from [36], the author’s original work
Reduction in Activity of the Mesolimbic Dopaminergic Pathway Under Neuropathic Pain
Extracellular signal-regulated kinase (ERK) is a serine/threonine kinase that mediates cellular responses to a wide variety of signals, including opioid receptor-regulated signaling [41, 42]. ERK activity in the VTA is increased by the chronic administration of morphine, and ERK activity in the VTA is associated with a morphine-induced increase in tyrosine hydroxylase (TH) [43], which is the rate-limiting enzyme in the biosynthesis of dopamine. Therefore, we examined whether ERK contributed to the rewarding effects of morphine and whether neuropathic pain could influence ERK activity in the mouse VTA [10]. Ligation of the sciatic nerve reduced the level of phosphorylated-ERK (p-ERK) in the VTA without any changes in the basal protein level of ERK. Furthermore, p-ERK immunoreactivity in the VTA that was mostly localized within TH-positive neurons was markedly decreased after sciatic nerve ligation [10]. Various protein kinases can phosphorylate TH at specific serine residues. ERK has been reported to phosphorylate TH at Ser31 in vitro [44]. Ligation of the sciatic nerve reduced p-TH (ser31)-immunoreactivity in VTA neurons that project to the NAc. [36]. These findings suggest that a persistent decrease in ERK activity in the VTA under neuropathic pain may reduce TH activity and result in reduced dopaminergic tone associated with potential dysphoria. This may in turn make the mesolimbic dopaminergic system less responsive to exogenously administered μ-agonists under neuropathic pain.
Related posts:
 Biopharmaceutical Considerations of Opioid Analgesics in Models of Self-Administration: Review and Summary
Prenatal Exposure to Opioids
Opioid and Non-Opioid Drug Responding Under States of Chronic Pain: A Timeline Spanning 1980 to Present Day
Opioid Self-Administration in the Presence of Chronic Pain: Analgesia or Addiction?
Opioids in an Evidence-Based World
Biopharmaceutical Considerations of Opioid Analgesics in Models of Self-Administration: Review and Summary
Prenatal Exposure to Opioids
Opioid and Non-Opioid Drug Responding Under States of Chronic Pain: A Timeline Spanning 1980 to Present Day
Opioid Self-Administration in the Presence of Chronic Pain: Analgesia or Addiction?
Opioids in an Evidence-Based World
 Drug Addiction and Chronic Pain: A Review of Animal Models
Drug Addiction and Chronic Pain: A Review of Animal Models
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


