McKee’s four stages of CTE tau pathology and associated clinical features. The left column depicts 50 μm coronal tissue sections with CP-13 immunostain which labels phosphorylated tau with a brown pigment. The middle column describes the anatomic distribution of tau. The right column describes the clinical features that were reported in association with each neuropathological stage of CTE.
No consensus has been reached on appropriate clinical diagnostic criteria for CTE. Criteria have been offered [35], but they emphasize clinical features that may be rare in modern CTE, such as cerebellar or pyramidal signs, are vague on the criteria requiring history of head injuries, and require features that are “clinically distinguishable from any known disease process,” an unrealistic expectation at this time in which distinguishing features of CTE are still largely unknown and most features described to date overlap with other known disease processes. While sensitive and specific diagnostic criteria will be valuable in furthering CTE research, these efforts are hampered by current limitations in knowledge about the illness, including better information about its longitudinal course and a better understanding of the magnitude of risk conferred by single or multiple TBIs or subconcussive injuries.
Epidemiology
TBI and risk of neurodegenerative disease
An accurate understanding of the risk conferred by TBI has been difficult to obtain. Because CTE is currently a pathological diagnosis, and autopsy series have inherent acquisition bias, there is no current way to define the prevalence in the population. Even within a limited group of athletes it is not possible to determine the prevalence of CTE or risk to an individual. A large, recent autopsy series that included 34 American football players found that the pathological stage of disease correlated with length of playing career [12], though this is an indirect measure for quantifying cumulative head injury. Questionnaires given to retired NFL players have found that the frequency of clinically diagnosed mild cognitive impairment, memory problems, and depression is elevated, particularly for those who reported three or more concussions compared to those who did not report a concussion history [30, 31]. A study of death certificates among retired NFL players found a three-fold elevated rate of dying from neurodegenerative causes compared to the typical population frequency, with AD and ALS particularly over-represented [36]. On the other hand, a study of those who played high school American football in Rochester, Minnesota between 1946 and 1956 found no increased rate of later developing dementia, PD, or ALS [37]. This latter study, however, must be interpreted in the context of dramatic changes in protective gear and body habitus of American football players, even at the high school level, over the past 60 years that may influence the biomechanical features of impacts. Prospective, longitudinal studies will be needed to better gauge the risk among American football players. Studies estimating the prevalence among other athletes have also been problematic. A 1969 study of retired boxers (including many who had fought in bareknuckle, less regulated fights) found that 17% had neurologic deficits that could be attributed to boxing [38]. Current longitudinal studies of boxers are underway [39]. The prevalence of PD among retired Thai traditional boxers is estimated at 0.71%, which is thought to be elevated relative to the population, and having > 100 bouts is an associated risk factor [40]. The initial screening for parkinsonism in this study was dependent on response to surveys, rather than uniform neurological examination.
Outside of athletics the effect of TBI on rates of neurologic and psychiatric illness has been the subject of many studies, but limitations in these have led to a lack of confidence in their findings. Meta-analyses have shown an increased risk associated with history of TBI for the development of AD [5, 41], PD [42], and ALS [10], though results and methodologies have varied widely among studies. TBI has been shown to be a risk factor for FTD [6, 43]. Psychiatric symptoms, particularly mood disorders, are common following TBI, but the risk of developing protracted psychiatric illness is less clear [44]. Many of these studies involved retrospective determination of the TBI, introducing recall bias and incomplete information about its timing and severity. Longitudinal cohort studies eliminate this recall bias, but often determine the presence of TBI by review of medical records or diagnostic codes, likely missing milder or repeated injuries, which the study of American football players suggests may be highly relevant. The timing of injury relative to the neurologic diagnosis is also critical, and often poorly assessed in prior studies. For example, an individual with PD may report a head injury that actually occurred due to the very early stages of the disease, rather than being causative, a phenomenon known to epidemiologists as “reverse causality.” In one study that stratified the interval between injury and diagnosis the risk of TBI on PD was no longer present when looking at injuries that occurred more than 10 years before diagnosis [45].
Additional risk factors
Apolipoprotein E (APOE) ε4 allele, the strongest susceptibility gene for AD, is associated with increased risk for many neurodegenerative diseases following TBI [4, 11, 34, 46]. This association, however, has recently been called into question with respect to CTE [12]. Exciting work in mouse models, however, suggests that APOE ε4 may impair the brain’s overall ability to recover following injury [47]. Similarly, alterations in progranulin metabolism have been proposed to underlie increased risk of FTD after TBI [48]. Specific mutations in genes encoding α-synuclein have been associated with increased risk of PD after TBI [8] and this risk is augmented in a more than additive manner with exposure to paraquat-containing pesticides [49].
It is currently unknown to what degree, if any, recreational drug or steroid use, alcohol abuse, chronic psychiatric disease, and cardiovascular risk factors have on modifying risk of CTE, but many of these factors may have been over-represented in autopsy series of professional athletes with CTE that have been reported to date. There is some evidence that TBI in children or adolescents may be particularly morbid [50, 51], suggesting that there may be “critical periods” during which TBI may be more likely to result in a progressive neurodegenerative cascade. Gender effects are also understudied as the vast majority of cases of CTE have been in males, presumably due to referral bias.
Neuroimaging and other biomarkers
There is a paucity of neuroimaging and biomarker data in cases of pathologically confirmed CTE. Most of our knowledge in this area is based on neuroimaging studies of concussed athletes and military veterans considered to be at risk for CTE. The importance of neuroimaging and other biomarkers in the assessment of CTE is manifold: to assist prognostication of TBI-exposed patients prior to development of frank CTE neuropathology, to assist in differential diagnosis of suspected cases of CTE, and to clarify neuroanatomical substrates of clinical features of CTE. Once effective therapies are developed, neuroimaging and other biomarkers will likely guide the choice of treatment in TBI-exposed patients.
Magnetic resonance imaging (MRI)
Autopsy studies have identified a number of gross pathological features associated with CTE, described in further detail in the neuropathology section of this chapter, which would likely be identifiable on structural brain imaging. Some of these features, including cavum septum pellucidum (CSP) [52–55] (Figure 23.2), hippocampal atrophy [55, 56], lateral ventricular enlargement [57], global cerebral atrophy [53, 58], and dilated perivascular spaces [55, 59], have been reported to occur at high rates in imaging series of boxers, other unarmed combatants, and civilians exposed to TBI. Among unarmed combatants, fighting exposure was found to significantly correlate with ventricular size and presence of perivascular spaces [55]. Many have hypothesized that the CSP seen in contact-sport athletes may result from a fluid-wave generated within the lateral ventricles that disrupts the septum pellucidum at the time of a traumatic hit [21, 60], while others have proposed that CSP in boxers is a congenital finding associated with the tendency of a person to choose to engage in boxing [61]. The identification of high rates of CSP in an autopsy series of victims of fatal motor vehicle accidents [62], however, renders this latter explanation less plausible.
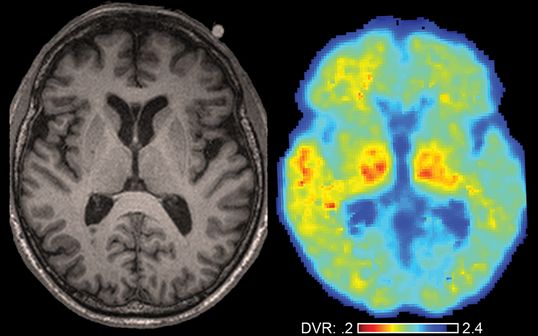
Neuroimaging in a retired American football player at risk for CTE. Axial T1 MRI (left) and axial PIB PET (right) of a 68-year-old retired professional American football player who presented to our center with short-term memory loss, irritability, insomnia, and tremor. MRI reveals a cavum septum pellucidum. PIB PET reveals focal amyloid deposition (in red) in the left anterior temporal lobe only, suggesting a traumatic etiology. Signal in thalami is considered artifactual. Right brain is shown on the right. Abbreviation: DVR = distribution volume ratio.
Given the frequent perivascular tau accumulation reported in CTE, it is hypothesized that vascular injury may play a role in the pathophysiology of CTE and thus may be a helpful neuroimaging marker. One study of retired professional American football players with cognitive complaints revealed increased white matter lesion volume on MRI fluid-attenuated inversion recovery (FLAIR) sequences, suggestive of increased microvascular lesions, compared to controls [63]. High-resolution susceptibility weighted imaging (SWI) may also have a role in identifying regions of parenchymal injury by highlighting micro-hemorrhages after acute TBI. It is currently unknown whether MRI FLAIR or SWI abnormalities, potentially indicative of static TBI-associated parenchymal injury, will prove useful for long-term prognostication or risk-stratification [64, 65].
Functional MRI has detected impairment in activation of dorsolateral prefrontal cortex in high school American football players subjected to repeated helmet collisions regardless of whether they demonstrated overt clinical symptoms of concussion [66]. Whether these players will be at increased risk of CTE compared to players without functional MRI changes is currently unknown. Functional MRI has also detected reductions in medial temporal lobe and inferior parietal lobe function in retired professional American football players that correlated with number of reported concussions [67]. Preliminary work using resting-state functional MRI suggests that TBI-mechanism (blast versus impact) may dictate the pattern of connectivity reductions, thus highlighting both the benefits and limitations of most imaging studies to date that have included populations of patients with fairly homogeneous TBI mechanisms [68]. Overall, these finding suggest that functional MRI may prove useful for risk-stratification in longitudinal studies.
Diffusion tensor imaging (DTI)
Similar to the findings reported in functional MRI, many DTI studies have detected changes after both recent and distant TBI exposure. DTI appears to be particularly sensitive to detecting diffuse axonal injury after mild TBI [69]. An elegant prospective study that objectively measured TBI exposure using instrumented helmets in collegiate athletes over a single season demonstrated correlation of head impacts with reductions in white matter diffusivity in corpus callosum, amygdala, cerebellum, hippocampus, and thalamus that correlated with worse performance on cognitive testing [70]. Widespread changes in DTI measures, including in corpus callosum and multiple other longitudinal and cortical–subcortical white matter tracts, in blast-exposed Iraq and Afghanistan war veterans have been reported that may be dose-dependent [71]. DTI has also been applied to retired professional NFL players in whom reduced integrity of the forceps minor has been reported in depressed versus non-depressed players [72] and in whom widespread reductions in fractional anisotropy throughout frontal and parietal regions have been identified in symptomatic retired players versus healthy controls [63]. Other preliminary work has reported abnormal thinning of colossal fibers in a small cohort of retired NFL players [73]. This work suggests that DTI may be a sensitive biomarker of both recent and distant TBI-related white-matter pathology, but further research with autopsy confirmation is needed to determine the utility of this modality in diagnosis or prognostication of CTE.
Positron emission tomography (PET)
PET holds great promise as a biomarker for the tau pathology associated with CTE. Tau-specific ligands are currently under development and are beginning to enter clinical trials [74–76]. The ligand 2-(1-(6-[(2-[F-18]fluoroethyl)(methyl)amino]-2-naphthyl)ethylidene)malononitrile or “FDDNP” has been touted by some as a marker of tau [77, 78] but it is unfortunately a non-specific marker of both amyloid and tau and thus may be less useful for establishing a diagnosis of CTE [73].
Cerebrospinal fluid (CSF) and blood biomarkers
As CSF is in direct contact with brain tissue, it presents a unique opportunity to measure byproducts of TBI and neurodegeneration. Levels of neurofilament light chain (and to a lesser degree, total tau) have been found to be increased in amateur boxers following a bout, to decline after 3 months of rest, and, in the case of neurofilament light chain, to correlate with the severity of head blows [79, 80]. Perhaps most promising, however, are blood biomarkers including neuron-specific enolase (a biomarker of neuronal damage) and S-100beta (a biomarker of glial damage) that have both been found to rise in response to head blows incurred by amateur boxers [81, 82], and in the case of neuron-specific enolase, to remain elevated even after an extended period of rest [81]. These biomarkers hold promise for use as diagnostic adjuncts or prognostic tools. Further work in this field is warranted to determine whether abnormalities in CSF or blood biomarkers may be detected even many decades after repetitive TBI or may herald the onset of CTE neuropathology.
Neuropathology
All autopsy-proven cases of CTE described in the literature to date have been in patients with a history of repeated TBI or subconcussive injuries including contact-sport athletes [12–16, 21, 32, 83], military veterans exposed to blast trauma [24, 84], and even civilians with a history of multiple falls or self-injurious head banging behavior [12]. While the predominant pathology in these cases is tau, modern staining techniques have also uncovered the presence of amyloid-β (Aβ) and transactivation responsive region de-oxyribonucleic acid-binding protein 43 (TDP-43) in many confirmed cases of CTE. Additionally, a significant minority of reported cases of CTE have also met neuropathological criteria for comorbid diagnoses of MND, PD, dementia with Lewy bodies, multiple system atrophy, AD, or frontotemporal lobar degeneration. Here we will focus on the neuropathological features that have been reported in “pure” CTE.
Gross pathology
Rarely, sequelae of a focal traumatic brain injury such as subdural hematoma may be seen [32]. Brains often show ventricular enlargement and cystic abnormalities of the septum pellucidum, such as CSP or cavum vergae, which appear to increase in frequency and size with increasing neuropathological grade of CTE. In more severe cases, the third ventricle may appear sharply concave and there may be atrophy of mammillary bodies, a convex contour of the medial thalamus, and thinning of the hypothalamic floor and corpus callosum. Grossly, depigmentation of locus ceruleus and substantia nigra may be visible. Ultimately, brain weight may be reduced and there may be gross evidence of frontal and temporal cortical atrophy, hippocampal atrophy or sclerosis, and white matter atrophy [12].
White matter and vascular injury
Axonal injury is uniformly present even in mild cases of CTE [12] and is hypothesized to underlie the early clinical symptoms of CTE at a time when tau pathology is sparse and unlikely to be causative. Sparse perivascular histiocytes, sometimes laden with hemosiderin, have been identified in many cases of CTE [32] which, along with the observation that the tau pathology appears to begin perivascularly in the depths of sulci [12], raises the possibility that microvascular injury may play a role in the pathophysiology of CTE.
Tau
Neocortical neurofibrillary cytoplasmic tangles, now known to contain aggregates of hyperphosphorylated tau, were first described in early reports of CTE in boxers [20] but have been subsequently confirmed by many independent groups [12, 32, 85–87]. The neurofibrillary tangles of CTE are described as band-shaped, flame-shaped, small globose, or large globose [32]. Additional tau pathology more recently identified in modern case series of CTE includes neuropil threads and astrocytic tangles [21, 32]. While neocortical tangles of AD show many similarities to those seen in CTE, such as the specific tau isoforms and ratio of 3-repeat to 4-repeat tau [87], the tangles in CTE have many distinguishing features such as their microscopic and macroscopic distribution [12, 21, 88, 89] (Table 23.1). In the most comprehensive modern autopsy series of CTE to date, McKee et al. [12] describe progressive stages of CTE pathology (Figure 23.1) that appears to begin perivascularly in the depths of sulci of the frontal lobes and then advance to widespread cortical and subcortical areas with an irregular patchy distribution (sometimes referred to as the “skip phenomenon” [32]), ultimately prominently affecting thalamus, hypothalamus, mammillary bodies, amygdala, substantia nigra, brainstem, white matter tracts, and even spinal cord. Others have described neuropathological phenotypes of CTE based upon cortical versus brainstem predominant distribution of tau pathology and presence or absence of Aβ co-pathology [32].
| AD | CTE | |
|---|---|---|
| Tau isoforms | All 6 isoforms, 3R and 4R | All 6 isoforms, 3R and 4R |
| Cells affected | N | N & A |
| Cellular tangle distribution | Laminae III & V (N) | Laminae II & III (N) Perivascular (N & A) Depths of cerebral sulci (N & A) Subpial and periventricular (A) |
| Gross tangle distribution | Braak stages [119, 120]. White matter tracts relatively spared even in later stages | Irregular patchy cortical distribution. White matter tracts prominently affected in later stages. Calcarine cortex usually spared |
Abbreviations: A = astrocytic, N = neuronal, 3R = 3 repeat, 4R = 4 repeat.
Amyloid-β
Once Aβ staining techniques were developed, a re-examination of the original CTE neuropathological case series of boxers [20] discovered extensive Aβ pathology in the cases with the most severe tau pathology [90]. More recently, Aβ has been identified predominantly as diffuse plaques, and only rarely as neuritic plaques or vascular amyloid, in a significant minority of cases of CTE not considered to meet neuropathological criteria for comorbid AD [12, 21]. This is in contrast to AD pathology in which Aβ is typically in the form of abundant dense neuritic plaques [91]. Interestingly, animal and human studies have discovered up-regulation of amyloid precursor protein (APP) after acute TBI [92–94], which is so reliable that APP is now widely used an immunostain marker of diffuse axonal injury (DAI) in humans [95]. Furthermore, up to 30% of patients who die within 1 hour of a severe TBI are found to have Aβ plaques on autopsy [92, 94].
TDP-43
While TDP-43 was originally considered a unique feature of FTD and ALS, it is becoming increasingly recognized as a feature of other neurodegenerative diseases including AD and CTE [96, 97]. In CTE autopsy series that have stained for TDP-43, it has been reported in over 80% of cases and is present even in cases without evidence of MND [12]. There is some evidence that when TDP-43 extends into anterior horn cells in patients with CTE, then the patient is likely to manifest clinical signs of MND, now referred to as CTE-MND [34]. Given the frequent and widespread co-occurrence of two abnormal aggregated phosphorylated proteins – TDP-43 and tau – in the vast majority of cases of CTE, a shared pathophysiological pathway seems plausible.
Pathophysiology
Mechanical trauma
The pathophysiological cascade leading to the development of CTE is believed to begin with exposure to repetitive non-penetrating head injury resulting in transmission of acceleration–deceleration biomechanical forces directly to neural tissue. Given reports of pathologically proven CTE in contact-sport athletes with a history of multiple helmet impacts without frank symptomatic concussions, it appears that even repetitive subconcussive injuries may be sufficient to trigger the development of CTE pathology [21, 60, 98]. The specific mechanism of injury likely dictates the location of brain tissue trauma. For example, rotational acceleration may produce diffuse axonal stretching and tearing, linear anterior–posterior acceleration may produce gliding contusions in parasagittal cortical regions and brainstem axonal stretch injury, and impact deceleration may produce coup–contrecoup cortical contusions particularly of basally or anteriorly located structures such as frontal or temporal poles or olfactory bulbs [35]. Furthermore, careful biomechanical evaluations of American football players wearing instrumented helmets reveal significant differences in frequency and magnitude of head impacts between different positions played, suggesting that variations may occur even among players of the same sport [99]. Animal models of blast trauma, on the other hand, found near instantaneous transmission of the unadulterated pressure wave directly to the entire brain that produced neuropathological changes of CTE after just a single exposure [24]. The accumulation of multiple repeated head impacts in all cases of CTE in humans, however, suggests that many may sustain multifocal cortical and white matter injuries.
Hypothetical neurodegenerative cascade
Animal models of white matter tract shear injury, or DAI, have shown immediate changes in axonal membrane permeability, massive calcium influx, and release of caspases and calpains, which have been implicated in subsequent tau phosphorylation and aggregation [100–103]. Once phosphorylated tau aggregates form, this may lead to a toxic gain of function that leads to continued phosphorylated tau aggregation and propagation via local extracellular non-synaptic spread as well as trans-synaptic spread [104, 105] to distant brain regions via functionally connected neuronal networks [106]. There is now human evidence for post-TBI axonal accumulation of tau as well as a variety of other abnormal proteins implicated in various neurodegenerative diseases including Aβ and α-synuclein [107], the latter of which has also been shown to spread from neuron to neuron [108, 109]. Propagation of these proteins in CTE could additionally be explained by cross-seeding in which accumulation of one abnormal protein enhances accumulation of either itself or a different abnormal protein via prion-like mechanisms [110, 111]. While these findings could explain the co-occurrence of multiple abnormal proteins in cases of CTE or, alternatively, lend support to the theory that TBI may lead to a number of divergent neurodegenerative conditions via a shared pathophysiological cascade, the duration of these acute post-TBI axonal processes is currently unknown. The long delay between TBI exposure and onset of clinical symptoms in most cases of CTE (or post-TBI cases of PD or AD, for that matter) suggests that the acute post-TBI neurodegenerative cascade either progresses extremely slowly or that the acute cascade may be independent from the later-onset cascade that ultimately leads to symptomatic neurodegenerative disease. Furthermore, the lack of CTE neuropathology in some patients with known exposure to repetitive TBI [112] suggests that there must be additional risk and protective factors that determine whether an individual exposed to TBI will recover uneventfully or trigger a progressive neurodegenerative cascade.
Reduced cognitive reserve
Aside from accumulation and propagation of abnormal proteins that may seed progressive neurodegenerative processes, a static axonal injury may simply lead to a reduction of cognitive reserve thereby increasing risk of subsequent dementia by accelerating onset of clinically apparent symptoms.
Prevention and treatment
Unlike most other neurodegenerative conditions, CTE has a clear environmental trigger: TBI. While TBI does not produce CTE in all exposed individuals, it appears to be a necessary trigger in all cases of CTE that have been described to date. There is mounting evidence that TBI also increases risk of a number of other neurodegenerative diseases. Thus, if TBI could be avoided, this would be expected to eradicate CTE entirely and may also reduce incidence of many other neurodegenerative diseases. Many TBIs sustained in the USA and globally, however, would be difficult to prevent given their accidental uncontrolled nature. TBI in the USA peaks in early childhood, adolescence, and again in later life with falls and motor vehicle accidents being the first and second leading causes, respectively [113, 114]. Thus, clinicians treating these patient populations are uniquely situated to intervene with primary preventative strategies to, for example, prevent falls in the elderly or prevent unrestrained motor vehicle accident injuries in children and adolescents.
TBI sustained in the controlled and highly regulated world of contact sports – from childhood to professional sports – present unique public health opportunities to make a substantial impact on reducing population risk of CTE in particular and neurodegenerative disease in general. While sports and medical associations have developed an array of post-concussion “return to play” guidelines [115–117], given the early state of science in this area, the details of these guidelines are often lacking good scientific bases [118]. At the moment, the most prudent approach is likely regular multimodality assessments to monitor for small deviations from baseline that may portend future progressive neurological decline. Longitudinal studies will help clarify the TBI and individual factors that exacerbate or mitigate CTE risk. As we advance our understanding of TBI-related neurodegenerative disease and improve our ability to monitor TBI-exposure and neurological sequelae, it will be increasingly incumbent upon those scientist and clinicians in the field to call for changes in public policy that will improve brain safety of individuals engaged in high-risk contact sports activities.
Currently, there are no disease-modifying treatments for CTE. Agents targeting tau are under development and may have a role in treating incipient or established cases of CTE. As the cascade of pathophysiological processes that occur from the moment of an acute TBI all the way to the delayed abnormal protein aggregation in CTE is further elucidated, this will pave the way for development of new therapies aimed at treating the spectrum of neurological injuries after TBI. Individuals suspected of having TBI-related neurological or psychiatric illness may benefit from treatments targeting their mood, headache, or other symptoms.
Summary
TBI is widely recognized as a risk factor for several neurodegenerative diseases including AD, FTD, PD, and ALS. Repeated mTBI or subconcussive injuries has also been associated with a unique tauopathy termed CTE that has, in postmortem analyses, been associated with cognitive, behavioral, and motor symptoms. Many, but not all, epidemiological studies support an association between TBI and later development of neurodegenerative disease. While all cases of autopsy-proven CTE to date have been in patients with a history of repeated mTBI or subconcussive injuries, many patients experience these injuries and do not develop CTE or other neurodegenerative diseases, indicating that many additional risk and protective factors, aside from TBI alone, must be important. Patients exposed to repeated mTBI or subconcussive injuries, such as athletes, show measurable changes on multimodal neuroimaging including structural MRI, functional MRI, and DTI that may be helpful for diagnosis, prognosis, and disease monitoring. CSF and serum biomarkers indicative of TBI or neurodegeneration may additionally be helpful. Neuropathologically, CTE is defined by neurofibrillary cytoplasmic tangles of hyperphosphorylated tau as well as frequent aggregates of TDP-43 and, occasionally, Aβ diffuse plaques. Animal and human studies support the triggering of a neurodegenerative cascade involving multiple abnormal proteins following TBI. Longitudinal studies including detailed clinical, neuroimaging, and biomarker assessments and ending with autopsy are needed to further define prevalence, clinical features, and natural history of CTE and other TBI-related neurodegenerative diseases and to pave the way for disease modifying treatment trials.
Acknowledgments
The authors have no disclosures. We thank Dr. Bruce Miller and Dr. Kristine Yaffe for their guidance and helpful comments. We thank Dr. Gil Rabinovici for providing a neuroimaging sample. We thank Dr. Anne McKee for providing photomicrographs. Lastly, we thank our patients and their families for contributing to research on traumatic brain injury and neurodegenerative disease.
References
Related posts:
 The non-fluent/agrammatic variant of primary progressive aphasia
The cognitive neurology of corticobasal degeneration and progressive supranuclear palsy
The non-fluent/agrammatic variant of primary progressive aphasia
The cognitive neurology of corticobasal degeneration and progressive supranuclear palsy
 Amnestic mild cognitive impairment
Amnestic mild cognitive impairment
 The Lewy body dementias
The Lewy body dementias
 Autoimmune antibody-associated encephalopathy and dementia syndromes
Autoimmune antibody-associated encephalopathy and dementia syndromes
 Sleep issues in dementia
Sleep issues in dementia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


