Clinical and Developmental Aspects of Pharmacokinetics and Drug Interactions
Jessica R. Oesterheld
Richard I. Shader
Andrés Martin
Overview
In the first part of this chapter, the concept of pharmacokinetics is differentiated from pharmacodynamics, and basic clinical pharmacokinetic principles that are shared by children, adolescents, and adults are reviewed, including therapeutic drug monitoring. Next, the pharmacokinetic differences that distinguish pediatric populations from adults are examined, highlighting the ontogeny of individual P450 cytochromes (CYPs), intestinal and hepatic influx and efflux transporters, and UDP-glucuronylsyltransferases (UGTs). Finally, since one of the more clinically relevant applications of pharmacokinetic principles is in helping to understand (and at times to predict) important drug interactions, emphasis has been placed on presenting basic principles that underlie drug interactions and the real-world strategies to prevent them. This chapter serves as a complement to the following and related chapter, in which specific drug classes and individual agents are discussed in detail.
Pharmacokinetics and Pharmacodynamics
Pharmacokinetic principles relate to the handling and disposition of drugs within the body (i.e., those biological processes that lead to changes over time in drug concentration in body tissues and fluids). In general, drug concentration in a target organ determines how long a drug’s therapeutic and adverse effects will last. Changes during development in the processes of drug absorption, distribution, metabolism, and excretion may have an impact on the delivery of drug to target tissues. By contrast, pharmacodynamic principles are concerned with the biochemical and physiological effects of drugs at their effect sites, with their specific mechanisms of action. Stated succinctly, pharmacokinetics refers to what the body does to a drug and pharmacodynamics to what a drug does to the body (Figure 6.1.1.1). The effects of a medication may change during development, as brain regions or neurotransmitter systems develop and mature at different rates. These developmental changes in neurochemical systems (pharmacodynamic systems) can influence both therapeutic response and side effect profile. For example: 1) Compared to adults, adolescents have a higher risk of dystonic reactions to conventional antipsychotics agent (1,2); 2) prepubertal children appear to be at a higher risk for the activating side effects of the SSRIs (3), and 3) developmental differences in the maturation of noradrenergic pathways may explain, at least in part, why tricyclic antidepressants are less effective in children with depression as compared to adults (4). Taken together, these findings suggest that major neurochemical systems that are altered by psychotropic drug treatments (e.g., dopaminergic, serotonergic, and noradrenergic, respectively) are subject to age-related effects.
Basic Principles
An understanding of pharmacokinetic principles is important for safe and effective patient care. Pharmacokinetic factors are often critical in a variety of clinical decisions, such as choosing between agents within a same drug class, switching between different medication preparations, adjusting dosages, preventing drug interactions, or correctly utilizing and interpreting therapeutic drug levels.
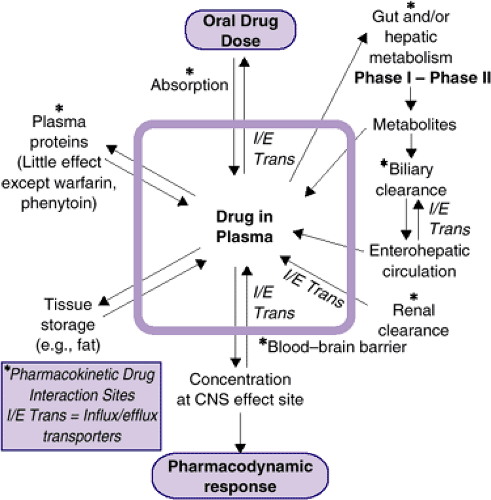 FIGURE 6.1.1.1. Pharmacokinetics and pharmacodynamics of a CNS drug. |
Pharmacokinetics can be conceptualized as having four functionally distinct phases: absorption, distribution, metabolism, and excretion. Absorption and distribution are primarily responsible for determining the speed of onset of drug effect, while the processes of metabolism and excretion terminate the action of the pharmacologic agent by removing the active form of the drug from the body. Taken together, these four phases help to determine the duration of drug activity (5).
Once a drug gains entry to the bloodstream, it is diluted in the plasma and bound at varying degrees to plasma proteins. The drug, usually protein-bound, is then either excreted by the kidneys or carried to the liver and transformed to a more water-soluble (and usually inactive) metabolite, which can then be excreted in urine or bile. This complicated and interdependent series of events is designed to reduce the effect of foreign molecules, with the ultimate goal of eliminating the drug from the body (Figure 6.1.1.1). If the dose of a drug is sufficiently large to withstand this “pharmacokinetic assault,” then a fraction of a psychoactive drug will cross the blood–brain barrier and endure to produce its pharmacodynamic effect (5). The last step of this process presumes that the drug in question is not a substrate for blood–brain barrier transport proteins that keep certain entities from reaching the brain.
Linear and Non-linear Pharmacokinetics
A basic working knowledge of key pharmacokinetics principles is relevant to the clinical practice of pediatric psychopharmacology to understand the fate of administered drugs. Some psychotropic medications follow first-order (or linear) kinetics, in which the amount of drug eliminated is proportional to its amount circulating in the bloodstream. Once alterations during absorption have taken place, first-order kinetics provide close to a one-to-one relationship between changes in dosage and in plasma concentration. Such a linear association generally allows for clinically relevant predictions of the impact of a dose change on circulating drug levels [bupropion SR (6), oxcarbazepine (7).] By contrast, zero-order (or non-linear) kinetics prevail when metabolizing or eliminating mechanisms are exceeded or saturated. This results in a fixed amount of drug being eliminated per unit of time, regardless of the plasma level. Certain drugs [paroxetine (8), nefazodone (9)] demonstrate zero-order kinetics at clinically relevant doses, making the relationship between dose changes and subsequent plasma levels much less predictable.
Vignette 1
A 12-year-old boy with a diagnosis of obsessive-compulsive disorder is not responding to 10 mg of paroxetine. Would an increase to 20 mg be appropriate? When the dosing of paroxetine is increased from 10 mg to 20 mg per day in children and teens, the plasma concentration can increase nearly seven-fold instead of a predicted two-fold if the drug’s kinetics were linear (8). Paroxetine inhibits its own catabolic pathway (CYP 2D6) and interferes with its own elimination. It could be prudent to increase the dose to 12.5 mg per day.
Multiple Dosing to Average Steady State Concentration
For drugs that follow first-order kinetics, the concepts of elimination half-life and of steady-state concentration are relevant for the practicing clinician. The elimination half-life (t1/2) is the time required for the concentration of drug to decrease by one-half. This term is also referred to as the beta-phase half-life or the biologic half-life. We prefer the term elimination half-life since it makes this concept distinct from the half-life of absorption and the half-life of biologic activity. In clinical practice, this parameter is usually assessed by measuring the decay of plasma or serum drug concentration and is referred to as the plasma or serum half-life. Plasma half-life values can be useful when determining dosing intervals. At consistent dosing intervals, it is the plasma half-life that determines the average plasma steady-state concentration (CSS). Css is reached when there is an equilibrium between the amount of drug ingested and the amount of drug eliminated, resulting in no net change in plasma concentration over time, and it is attained after four to five half-lives. The same time is necessary for reaching a new Css if daily dosing is increased or decreased or for complete elimination after drug intake is abruptly stopped (Figure 6.1.1.2). Most of the drugs commonly used in child psychiatry reach Css. However, in extensive CYP 2D6 metabolizers, neither psychostimulants nor atomoxetine does because of very short half-lives and therapeutic effectiveness at low concentrations. Carbamazepine, which induces its own metabolism, may take a much greater time to reach Css.
Vignette 2
A 14-year-old girl with major depression has failed a trial of fluoxetine, and she has been treated with 150 mg of twice-daily bupropion SR for 2 weeks. She has developed adverse cognitive effects. The clinician wishes to know if he discontinues the drug, how long before the adverse effects are likely to diminish. The half-life of bupropion SR in youth is approximately 12 hours (6), and in four to five half-lives or 2 to 3 days, it will be eliminated. This is an approximate answer because certain side effects may have a different concentration response relationship from the main therapeutic effects.
Therapeutic Drug Monitoring
Css is sometimes misunderstood by clinicians to imply an absence of daily peak and trough concentrations. Peak and trough concentrations exist within each dosing interval, and
when maximum and minimum drug concentrations are the same with two or more successive doses, Css is reached (Figure 6.1.1.2). Dosing some drugs once a day because they have a sufficiently long half-life may lead to excessive peak concentrations that can be associated with toxicity or increased adverse effects (e.g., clozapine is dosed twice daily because of potential for seizures at peak concentration). As Figure 6.1.1.3 shows, if the initial dose is doubled and dosed once daily, the peak concentration exceeds the desired concentration to toxicity, but if the original dose is doubled but given twice daily, the concentration is above the plasma concentration that is likely to produce a clinical effect, the minimal effective concentration (MEC), but below toxic levels, the maximal tolerable concentration.
when maximum and minimum drug concentrations are the same with two or more successive doses, Css is reached (Figure 6.1.1.2). Dosing some drugs once a day because they have a sufficiently long half-life may lead to excessive peak concentrations that can be associated with toxicity or increased adverse effects (e.g., clozapine is dosed twice daily because of potential for seizures at peak concentration). As Figure 6.1.1.3 shows, if the initial dose is doubled and dosed once daily, the peak concentration exceeds the desired concentration to toxicity, but if the original dose is doubled but given twice daily, the concentration is above the plasma concentration that is likely to produce a clinical effect, the minimal effective concentration (MEC), but below toxic levels, the maximal tolerable concentration.
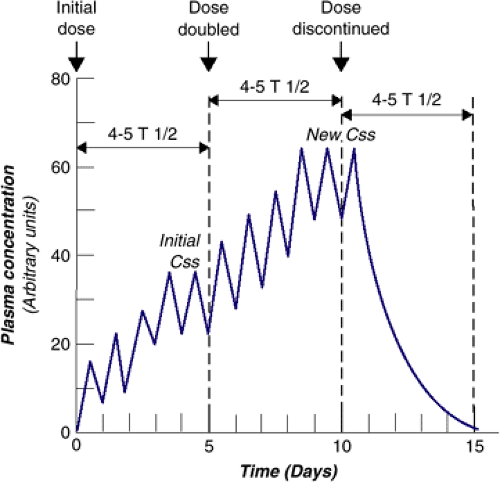 FIGURE 6.1.1.2. Multiple drug dosing leads to steady-state concentration. |
Clinicians must tailor drug dosing and frequency to maximize the probability of drug efficacy and to minimize toxicity. Some drugs (lithium, valproate, carbamazepine, nortriptyline) that have narrow therapeutic indices, significant consequences associated with toxicity, wide interpatient variability, and no clinical endpoint to guide drug dosing, are candidates for therapeutic drug monitoring (TDM). A final requirement must be met: The concentration of a drug must have a proportional relation to the concentration and the pharmacologic action at the receptor site (10). Ranges of concentrations have been established for drugs that meet these requirements, and values for psychotropic drugs used by child psychiatrists are given in Chapter 6.1.2.
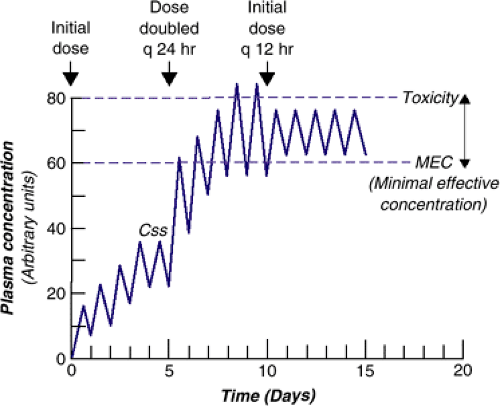 FIGURE 6.1.1.3. Changes in plasma concentration with changes in amount of drug or frequency of dosing. |
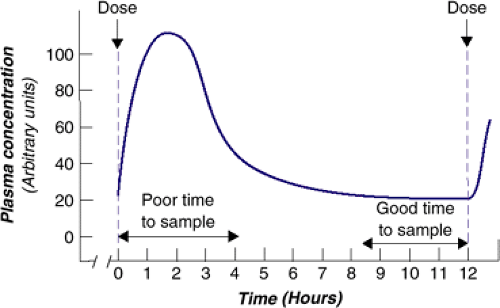 FIGURE 6.1.1.4. Idealized plasma curve. |
The range between subtherapeutic and toxic doses, or therapeutic range (sometimes called the therapeutic window) is a misnomer of sorts, since even when drug concentration is within the range, not every youth will respond to the drug, and a percentage will experience toxicity. For some drugs, there are data only on the MEC. Therapeutic drug monitoring (TDM) can reveal individuals with unusual metabolism (see the metabolism section), uncover nonadherance, demonstrate increased or decreased concentration with the addition of other drugs (see drug interaction section), and confirm toxicity. TDM provides information that supplements clinical assessment.
There are two common errors in using TDM: not waiting for Css to occur before drawing a blood sample, and not drawing the blood sample at trough level.
As Figure 6.1.1.4 shows, sampling at “trough,” or just before the next dose, offers the flattest part of the curve. For drugs given two or three times daily, sampling blood concentration before the AM dose in the morning is recommended, and for drugs given once daily, sampling before the next dose is suggested.
Vignette 3
A 17-year-old girl with bipolar disorder has been maintained on 600 mg of lithium carbonate twice daily. A trough level of lithium of 0.8 mEq/l has been maintained for 8 months, with reduction in her symptoms. Parents phone the clinician because their daughter is nauseated and vomiting. As previously instructed, the teen has not been given her morning medication. The finding of a trough level of 1.4 mEq/l initiates careful inquiry and reveals that friends have been giving her ibuprofen for menstrual cramps. By inhibiting prostaglandins, NSAIDs affect renal blood flow and thus the renal clearance of lithium (11).
Pharmacokinetics in Children and Adolescents
There are many pharmacokinetic similarities between adults and children and adolescents. Indeed, age-independent genetic influences on metabolism can be more salient than those influences attributable to age and developmental change. Nonetheless, children and adolescents do display unique pharmacokinetic parameters. Premature infants, neonates, toddlers, children, and adolescents are not a homogenous group in terms of drug distribution patterns (12). These differences can
be especially dramatic both at the neonatal stage and around the time of puberty, when the release of gonadal hormones can strongly influence plasma drug concentrations (13).
be especially dramatic both at the neonatal stage and around the time of puberty, when the release of gonadal hormones can strongly influence plasma drug concentrations (13).
Factors Affecting Drug Disposition
Absorption and Bioavailability
Drugs gain entry into the body through a variety of portals. The bioavailability of a medication in the systemic circulation— the amount of unbound drug available to exert a biological effect on target tissues— is determined by its absorption and, for orally administered medications, by presystemic clearance (first pass effect) from intestinal and hepatic transporters, metabolism, and conjugation (see next section) and protein binding. Drugs have variable first pass effects. Some drugs are metabolized very efficiently on first pass (60 to 70%) and others less so (less than 30%). Bioavailability measures the completeness of drug absorption, and it is determined in reference to intravenous dosing. Oral administration is by far the most common portal of entry but often the most unpredictable in terms of final bioavailability. Some drugs are given to children as tinctures or in alcohol-based syrups. Although drugs like penicillin G are absorbed in the stomach, most psychotropic medications are absorbed in the proximal small intestine.
Little information is available regarding the effect of age on the absorption of psychotropic medications, although there are several theoretical considerations regarding the influence of this process in children and adolescents. A major factor influencing gastrointestinal absorption is pH-dependent diffusion. In infants, the gastric pH is nearly neutral in the first week after birth. It slowly reaches adult values by age three years (14). In toddlers, stomach contents tend to be less acidic than in adults, causing weakly acidic drugs to be more highly ionized. Because it is the un-ionized fraction that is absorbed from the stomach, weakly acidic drugs may be absorbed more slowly in children. This process theoretically could affect phenobarbital and other anticonvulsants, amphetamines, and antidepressants (15).
Other factors that could reduce overall absorption are gastric and intestinal transit time. Gastric transit time is likely increased in neonates and infants, and the age of maturation is not known (14). Intestinal transit time is increased and the absorptive surface area of the intestine is reduced in young children, suggesting that drugs with a long phase of absorption (e.g., carbamazepine) and some sustained-release preparations may be incompletely absorbed (16). It is important to remember that although the rate at which many drugs are absorbed is slower in neonates and infants, there are no data indicating a generally reduced absorption of orally administered drugs in prepubertal children or teenagers (17).
Intestinal Influx and Efflux Transporters and Intestinal Metabolism
Until 10 to 15 years ago, it was believed that the first-pass effect was limited to the hepatic metabolic enzymes and conjugation, and that diffusion was the only mechanism involved in absorption in the small intestine. It was known that lipid-soluble drugs passively diffuse through the apical membrane of the small intestine, that some small hydrophilic ionized drugs squeeze through intracellular junctions, and that both types of drugs cross the cytosol and exit the basolateral membrane into the portal circulation (18). It is now known that non–lipid-soluble drugs are also actively transported (both imported and exported) across these liminal boundaries by members of the solute carrier family of transporter proteins that currently includes 43 subfamilies and 298 transporter genes (19,20) (Figure 6.1.1.5). Several names exist for each transporter especially in the earlier literature.
Best characterized of these influx transporters is the peptide transporter 1, PEPT1, SLC15A1. This transporter uses the intestinal-cellular proton gradient as a source of energy to ferry dipeptides and tripeptides across the apical membrane. Hundreds of peptides and other molecules are possible substrates (beta-lactam antibiotics, ACE inhibitors, thrombin inhibitors, acyclovir, sulpiride, and others). After crossing to the cytosol, these drugs are shepherded across to the portal circulation by a second peptide transporter embedded in the basolateral membrane (21) (Figure 6.1.1.5). Although the development of SLC15A1 has been studied in mice and rats, there is no information about when it matures in humans. Targeting the peptide transport system represents a new strategy for drug delivery of poorly absorbed drugs. Acyclovir, a polar antiviral drug, is poorly absorbed from the intestine, but if the L-valyl ester pro-drug valacyclovir is given, it is transported by SLC15A1, and therapeutic levels can be obtained. The ester drugs like methylphenidate are also hydrolyzed by plasma esterases. In the case of methylphenidate, the inactive metabolite ritalinic acid is formed. In the case of acyclovir, an active metabolite is formed.
Efflux transporters are also embedded in both of the intestinal membranes. Members of a superfamily of transporters that use ATP as an energy source (ATP-binding cassette transporters or ABC transporters (20) can flip compounds which have entered the cytosol back into the intestinal lumen on the apical side and from the cytosol into the portal system on the basolateral side (Figure 6.1.1.5). Efflux transporters act not only to limit drug absorption and bioavailability in the intestine, but they efflux compounds into the biliary system and the kidney, protect “sanctuaries” such as the brain (Figure 6.1.1.1), and are responsible in part for resistance to cancer drugs.
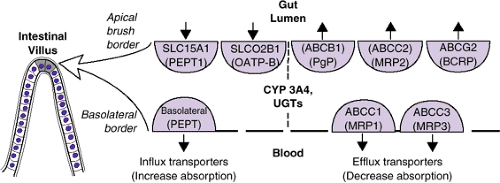 FIGURE 6.1.1.5. Intestinal transporters. |
About 30 years ago, Juliano and Ling (22) noted that after initial efficacy, many oncologic drugs stopped being effective at the same time; they named this phenomenon multiple drug resistance (MDR). A gene on chromosome seven (MDR1) was found to encode a glycoprotein (P-glycoprotein, P-gp) that pumps out compounds from cells. Multiple drugs were affected at the same time because all of them were P-gp substrates.
P-gp is now numbered 1 of subfamily B of superfamily ABC (ABCB1). Other members of this family involved in efflux drug transport are in subfamily C (Figure 6.1.1.5). There is an avalanche of in vitro data on what compounds are substrates of these transporters, and how other drugs, foods, or genetic variations can affect ABCB1. Since ABCB1 is saturable in vivo, only drugs that are therapeutic in low doses can be shown to be affected [digoxin, talinolol, fexofenadine, (23)]. Drugs that affect ABCB1 either through inhibition or induction represent a new form of drug interaction. For example, quinidine, an inhibitor of ABCB1, blocks the efflux of digoxin in the kidney, and more digoxin will be absorbed and enter the circulation [(23); see drug interaction section].
In the intestine, both ABCB1 and CYP 3A4 are located in the endoplasmic reticulum. There is a considerable overlap in compounds that are substrates of both (dexamethasone, diltiazem, vincristine).
ABCB1 and CYP 3A4 act together as an intestinal defense tag-team to protect against exogenous compounds: ABCB1 provides a barrier to absorption and any remaining compound is converted by CYP 3A4 to less active metabolites [(24); see metabolism section and drug interaction section]. Levels of both ABCB1 and CYP 3A4 are higher in the intestine than in the liver, and there is a growing awareness of the importance of these intestinal proteins as compared to hepatic ABCB1/CYP 3A4 in first-pass metabolism. There is no coordination between the duodenal and the hepatic tag-teams, and in each organ there is a different ontogeny of CYPs and ABCB1 [(25); see metabolism section]. ABCB1 is also found in the blood–brain barrier, in the prostate, and in the placenta.
Drug Distribution and Plasma Proteins
Following absorption, drugs are distributed into intravascular and various extravascular spaces. Numerous physical factors can influence the distribution of a drug throughout the body: the size of body water compartments and adipose tissue depots, cardiac output, regional blood flow, organ perfusion pressure, permeability of cell membranes, acid-base balance, and binding to plasma and tissue proteins (13). Each of these factors may change during development, resulting in changes in the distribution of a drug and, subsequently, in its pharmacological effect.
Drugs are transported in the general circulation in two forms that are in dynamic equilibrium with each other: bound to plasma protein (acidic drugs to albumin and basic drugs to alpha 1-acid glycoproteins) and unbound (free). Only the unbound drug is usually available to pass across membranes and to have pharmacological effects. Although albumin and alpha 1-acid glycoproteins are reduced in the neonate and infant, this does not appear to be an important developmental factor in older children and adolescents (14,17).
Two important factors affecting distribution that change substantially during development are fat stores and the relative proportion of total body water to extracellular water. The relationship between the amount of drug absorbed (D), also referred to as the concentration at time zero, plasma concentration (Cp), and volume of distribution (Vd) can be summarized by the simple equation: Cp = D/Vd. Note that the larger the Vd, the smaller the Cp. The proportion of body fat is highest in the first year of life, followed by a steady decrease until an increase occurs prepubertally (12,26). The adipose tissue of infants has a lower ratio of lipid to water (17). The relative volume of extracellular water is high in children and tends to decrease with development. For example, total body water decreases gradually from about 85% of body weight in a small premature infant to about 70% in the full-term newborn to about 60% in the 1-year-old infant, a level that is generally maintained throughout adulthood. Similarly, extracellular water decreases gradually from about 40 to 50% of body weight in the newborn to about 15 to 20% by age 10 to 15 years (27). Thus, if weight-based drug administration is utilized, infants will tend to have lower drug plasma levels (17). Drugs that are primarily distributed in body water (lithium) can be expected to have a lower plasma concentration in the pediatric population compared with that in adults because the volume of distribution is higher in children and early adolescents.
Related posts:
 Child and Family Policy: A Role for Child Psychiatry and Allied Disciplines
Oppositional Defiant and Conduct Disorders
Psychodynamic Principles in Practice
Child and Family Policy: A Role for Child Psychiatry and Allied Disciplines
Oppositional Defiant and Conduct Disorders
Psychodynamic Principles in Practice
 Evidence-Based Practice as a Conceptual Framework
Suicidal Behavior in Children and Adolescents: Causes and Management
Integrating Behavioral Services into Pediatric Care Settings: Principles and Models
Evidence-Based Practice as a Conceptual Framework
Suicidal Behavior in Children and Adolescents: Causes and Management
Integrating Behavioral Services into Pediatric Care Settings: Principles and Models
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


