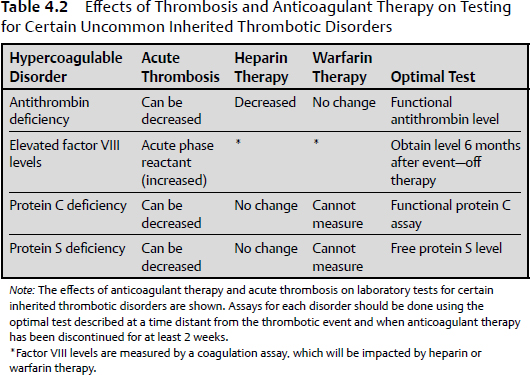
4 This chapter briefly summarizes disorders of bleeding and thrombosis, including both inherited and acquired etiologies. Acquired etiologies of bleeding and thrombosis are more common than the inherited forms. Thrombosis is the process of forming a blood clot within a blood vessel, thus obstructing the flow of blood through the circulatory system. Most commonly, presentations of thrombosis occur as deep venous thrombosis of the lower extremity with or without pulmonary embolism. The underlying cause can be classified as acquired or hereditary. Virchow’s triad proposes thrombosis to be a consequence of one or more of the following: alterations in blood flow, vascular endothelial injury, and alterations in the constituents of the blood. The discussion in this chapter focuses primarily on venous thromboembolism (VTE). Inherited thrombophilia is a genetic tendency for thrombosis, which typically presents in younger patients (< 50 years of age) and is usually recurrent in nature. The most frequent causes are the factor V Leiden and the prothrombin gene mutations, which account for about 40% of the cases. Table 4.1 summarizes the inherited states, their prevalence, and the associated increased risk for thrombosis. The effects of anticoagulant therapy and the setting of an acute thrombosis may both confound the laboratory evaluation of thrombophilia. However, the DNA tests for the factor V Leiden and prothrombin gene mutations, and an amino acid level for homocysteine, are not affected by anticoagulation and can be ordered at any time without concern about an incorrect test result. Table 4.2 summarizes the other inherited thrombophilias and potential confounding factors, which may affect a correct laboratory result. Although an extensive laboratory evaluation may identify an inherited thrombophilia in over 50% of affected patients, the available data indicate that positive thrombophilia test results do not change how patients should be managed.1 Table 4.1 The Inherited Hypercoagulable States
Clinical Disorders of Coagulation and Platelet Function
Thrombosis
Overview of the Causes of Venous Thrombosis
Inherited Thrombophilia
Risk Factor | Prevalence (Caucasians) | VTE Risk |
Factor V Leiden | Heterozygous 4% | 7× |
Prothrombin mutation | 2% | 2.8× |
Hyperhomocysteinemia | 5–10% | 2.5× |
Protein C or S deficiency | 0.3% | 10× |
Antithrombin deficiency | 0.04% | 25× |
Note: The six most common inherited thrombotic disorders are shown with their prevalence in Caucasians along with their associated VTE risk.
Acquired Venous Thrombophilia
Predisposing conditions for acquired venous thrombosis include a prior thrombotic event, recent major surgery or hospitalization, presence of a central venous catheter, trauma, immobilization, malignancy, pregnancy, certain medications (tamoxifen, lenalidomide), myeloproliferative disorders, and antiphospholipid antibodies. Many patients presenting with an episode of VTE have multiple risk factors. This was confirmed in a 1999 population-based study of VTE in Worcester, Massachusetts, in which six medical characteristics were identified2:
1. More than 48 hours of immobility in the preceding month (45% of patients)
2. Hospital admission in the past 3 months (39%)
3. Surgery in the past 3 months (34%)
4. Malignancy in the past 3 months (34%)
5. Infection in the past 3 months (34%)
6. Current hospitalization (26%)
Only 11% of the 587 episodes of VTE in this study had none of the six risk factors, whereas 36% had one or two and 53% had > 3 risk factors.
Malignancy
Cancer patients have an increased risk of thrombotic events due to tumor expression of procoagulants, such as tissue factor. Clinical VTE occurs in about 5% of cancer patients3 and in 12% of cancer patients with a central venous catheter.4
Surgery
Thrombotic risk is greatly increased during and after surgery. Additional risk factors in this group include older age, previous VTE, the coexistence of malignancy or medical illness, inherited thrombophilia, longer procedures, anesthesia, and longer immobilization times.5 Thromboprophylaxis greatly reduces the incidence of symptomatic VTE in the immediate postoperative period. There continues to be a risk for subsequent VTE following discharge, especially in orthopedic patients who require longer thromboprophylaxis.6 Neurosurgical patients specifically have a high incidence of VTE as shown in a prospective study done in 2009. Using mechanical prophylaxis, five of 37 patients (13.5%) developed asymptomatic DVT confirmed by ultrasound. Of those five patients, three eventually had a pulmonary embolism, suggesting the need for additional pharmacological prophylaxis.7
Thromboprophylactic management of the neurosurgical patient, with a high risk for both thrombosis and intracranial bleeds, was recently reviewed by Niemi and Armstrong.8 They concluded that thromboprophylaxis and bridging therapy should be tailored to the individual risks and the type of neurosurgery. The bleeding risk is minimized by allowing coagulation capacity to normalize preoperatively and by using reduced doses of low molecular weight heparin (LMWH) starting relatively late after neurosurgery.
Trauma
Major trauma significantly increases the risk of VTE. One study detected thrombi in 54% of patients with major head trauma, 61% of patients with pelvic fractures, 77% of patients with tibial fractures, and 80% of patients with a femoral neck fracture.9 Increased VTE risk has also been associated with a minor injury occurring in the preceding 3 to 4 weeks.10
Pregnancy
Estimates of the age-adjusted incidence of VTE range from 5 to 50 times higher in pregnant versus nonpregnant women and is likely due to obstruction of venous return from the enlarged uterus as well as the hypercoagulable state associated with pregnancy.
Drugs
Oral and transdermal contraceptives increase the risk of VTE within 4 months of the initiation of therapy. The Women’s Health Initiative along with the Heart and Estrogen/progestin Replacement Study (HERS) have also shown a twofold increase in VTE associated with hormone replacement therapy, which appeared greatest in the first year of treatment.
Immobilization
This category encompasses patients with recent hospitalizations; patients on bed rest; deconditioned elderly patients; patients with stroke, heart failure, or recent myocardial infarction; as well as patients who have undergone recent extended travel.
Antiphospholipid Antibodies
The antiphospholipid syndrome is characterized by the presence of antibodies directed against plasma proteins bound to anionic phospholipids in patients who present with arterial or venous thrombosis, complications of pregnancy, recurrent fetal loss, or thrombocytopenia. Etiologies may be either primary (idiopathic) or secondary due to autoimmune syndromes such as systemic lupus erythematosus, malignancy, infections, or drug reactions. Although a prolonged partial thromboplastin time (PTT) that does not correct with mixing with normal plasma may be seen, optimal testing for antiphospholipid antibodies includes assays for the lupus anticoagulant, immunoglobulin (Ig) G and IgM anticardiolipin antibodies, and IgG and IgM antibodies to β2-glycoprotein-1. In contrast to test results indicating inherited thrombophilia, which usually do not change patient management, positive results for antiphospholipid antibody tests do change the duration of treatment. Patients with antiphospholipid syndrome should receive therapeutic anticoagulation for as long as positive antiphospholipid antibody test results persist.11
Myeloproliferative Neoplasms and Paroxysmal Nocturnal Hemoglobinuria
The chronic myeloproliferative neoplasms, particularly polycythemia vera (PV) and essential thrombocythemia, are characterized by thrombotic complications, both arterial and venous. Paroxysmal nocturnal hemoglobinuria (PNH) is a clonal bone marrow disorder that results in intravascular hemolysis with episodes of hemoglobinuria and occasional leukopenia or thrombocytopenia. PNH is associated with an increased incidence of venous or arterial thrombosis.
Renal Disease
Chronic renal disease, nephrotic syndrome, and renal transplantation have all been reported to have an increased incidence of VTE. Patients with stage III/IV chronic renal disease have a relative VTE risk of 1.7 compared with patients with normal renal function.12
Chronic Liver Disease
Contrary to the popular belief that auto-anticoagulation occurs with liver disease and cirrhosis, a retrospective cohort of 190 hospitalized patients with chronic liver disease showed 12 (6.3%) developed VTE.13 The mechanism may be a result of acquired protein C and S deficiency.
Hyperviscosity
Thrombosis can be a manifestation of diseases associated with serum hyperviscosity (Waldenström macroglobulinemia or multiple myeloma), an increased number of red blood cells (PV), or a decrease in deformability of red cells as seen in sickle cell disease. Presenting symptoms include bleeding due to platelet dysfunction, visual disturbances, neurologic defects, deep venous thrombosis, pulmonary embolism, and portal and hepatic venous thrombosis.
Hyperhomocysteinemia
This disorder may occur either as a genetic or an acquired abnormality. The genetic disorder is associated with homozygosity for the thermolabile mutant of the enzyme methylenetetrahydrofolate reductase, or heterozygosity or homozygosity for cystathionine β-synthase. Homocysteine concentrations can also be elevated in acquired disorders such as vitamin B6, vitamin B12, and folic acid deficiencies.
Bleeding Disorders
Platelet Disorders
Disorders of platelet function include a common inherited bleeding disorder (von Willebrand disease), several rare congenital disorders, as well as a myriad of common acquired conditions. Platelet-type bleeding symptoms include easy bruising, mucocutaneous bleeding, and menorrhagia.
Acquired platelet disorders are more common, with the most likely etiology being secondary to therapeutic antiplatelet agents. These agents include aspirin, other nonaspirin nonsteroidal anti-inflammatory drugs, dipyridamole, clopidogrel, and other glycoprotein (GP) IIb/IIIa receptor antagonists, including abciximab and eptifibatide. Another acquired platelet function disorder is liver disease, which can induce both qualitative and quantitative platelet defects in not only chronic disease but also acute liver damage. Cardiopulmonary bypass also can cause significant platelet dysfunction due to numerous factors, including the interaction of platelets with the nonphysiologic surface components of the bypass membrane. Hypothermia during bypass, complement activation, release of cytokines, and thrombin generation may also contribute.14 Uremia associated with chronic renal failure has also been associated with increased clinical bleeding due to intrinsic platelet metabolic defects, and defects in platelet-endothelial interactions. Of course, malignancy and clonal disorders as well as thrombocytopenia secondary to several underlying disorders all may contribute to an increased propensity to bleed excessively.
Inherited disorders of platelet function include the most common bleeding disorder, von Willebrand disease (vWD), as well as the uncommon qualitative platelet disorders Bernard-Soulier and Glanzmann thrombasthenia, and disorders of platelet secretion and signal transduction including storage pool diseases, Hermansky-Pudlak syndrome, and Quebec platelet disorder. This category also includes signal transduction defects as well as abnormalities in arachidonic acid pathways and thromboxane A2 synthesis. Defects in cytoskeletal regulation include Wiskott-Aldrich syndrome. Finally, Scott’s syndrome is characterized by a defect in platelet procoagulant function.
Coagulation-Type Bleeding Disorders: Acquired and Hereditary
Coagulation-type bleeding symptoms include deep soft tissue hematomas, visceral bleeding, and hemarthrosis. The most common acquired coagulation disorder is associated with anticoagulant drugs, for example antithrombin inhibitors such as heparin products, factor Xa inhibitors such as fondaparinux, and the vitamin K antagonists such as warfarin. Liver disease and vitamin K deficiency from poor or inadequate nutrition can also lead to a coagulopathy. Acquired inhibitors, due to antibodies, can also either inhibit the activity or increase the clearance of a clotting factor, making a patient more likely to bleed. The most common antibodies that affect clotting factor activity are directed against factor VIII, in a disorder known as acquired hemophilia A. This condition can be seen in postpartum women, rheumatologic disease, and certain solid malignancies. Other inhibitors include antibodies directed against other coagulation proteins.
Inherited or congenital disorders of coagulation proteins include vWD, hemophilia A (factor VIII deficiency), and hemophilia B (factor IX deficiency), as well as less common factor deficiencies, such as fibrinogen, prothrombin, and factors V, VII, X, XI, and XIII. Laboratory evaluation of coagulation disorders is discussed in Chapter 3.
KEY POINTS
• Disorders of bleeding and thrombosis can be categorized as either acquired or inherited.
• Clinical history and appropriate laboratory testing determine whether a patient has either a platelet-type or a coagulation-type bleeding disorder.
• Inherited thrombophilia typically presents in patients younger than 50 years of age and is usually recurrent in nature.
• Acquired thrombosis is more common than the inherited thrombophilias, and may be seen in patients with any or all of the following: alterations in blood flow (stasis), vascular endothelial injury, and alterations in the constituents of the blood (hypercoagulability).
Related posts:
 Principles of Blood and Volume Replacement
Drugs Affecting Coagulation and Platelet Function
Principles of Blood and Volume Replacement
Drugs Affecting Coagulation and Platelet Function
 Intraoperative Non-Hematologic Adjuvant Methods for Preventing Blood Loss
Intraoperative Non-Hematologic Adjuvant Methods for Preventing Blood Loss
 Cranial Venous Sinus Thrombosis Diagnosis and Management
Cranial Venous Sinus Thrombosis Diagnosis and Management
 Neuroendovascular-Specific Patient/Case Examples
Neuroendovascular-Specific Patient/Case Examples
 Intraoperative Spine Surgery–Specific Patient/Case Examples
Intraoperative Spine Surgery–Specific Patient/Case Examples



