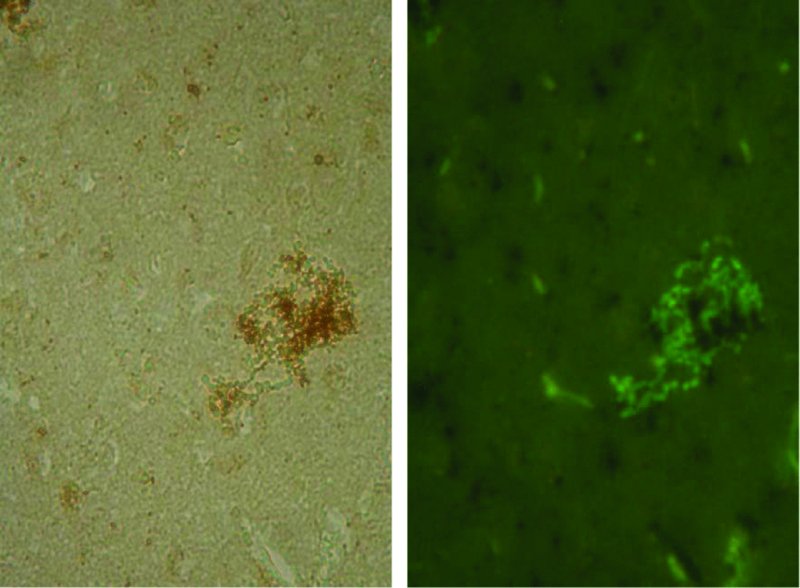
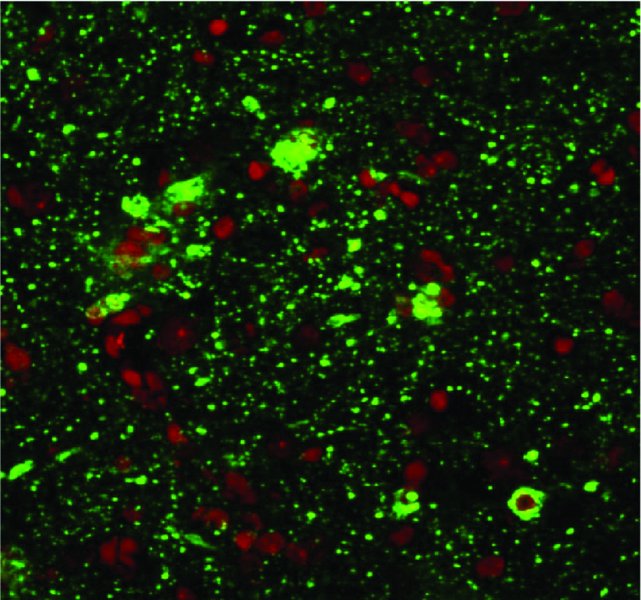
7 S. Louise Cosby1, Sareen Galbraith2 and Derek Healy3 1Centre for Infection and Immunity, School of Medicine, Dentistry and Biomedical Sciences, Medical Biology Centre, Queen’s University Belfast, Belfast, UK 2Department of Clinical Infection, Microbiology & Immunology, Institute of Infection & Global Health, University of Liverpool, Liverpool, UK 3Wildlife Zoonoses & Vector-borne Diseases Research Group, Animal Health & Veterinary Laboratories Agency, Weybridge, Surrey, UK Numerous microorganisms infect the human central nervous system (CNS). These encompass viral, bacterial, fungal and protozoan infections. In this chapter we focus primarily on neurological diseases caused by viruses and the virus-like agents, prions. Types of virus infection which occur in humans differ with both the geographical location and the immune status of the host. In Europe and North America, encephalitis due to viral infections is the predominant cause of CNS infection. Herpes viruses are the most common cause of CNS infection in temperate climates, but new and emerging infections which can result in CNS infection are spreading due to sociological factors, climate change and opportunity in immunosuppressed individuals. While poliovirus has been mainly eradicated from developed countries due to an effective vaccination policy (Minor, 2012), other CNS infections are becoming more frequent. Enterovirus 71 (EV71), in the same picornavirus family as polio, has emerged as an important human pathogen which may cause severe neurological complications and has caused death in children in outbreaks in the Asia–Pacific region during the past two decades (Solomon et al., 2010; Wilson, 2013). The World Health Organization has set regional elimination goals for measles virus (MV) eradication to be achieved by 2020 or earlier. However, there are concerns that zoonotic infection from the closely related and more neurovirulent veterinary morbilliviruses, particularly canine distemper virus, could occur (Cosby, 2012). Infections spread as a result of climate change by allowing the insect vectors to, for example, move northwards are exemplified by West Nile virus (WNV), Japanese encephalitis virus (JEV), the newly emerging Toscana virus and enterovirus-71 (EV71) (Charrel et al., 2005; Tyler, 2009; Wang et al., 2012). Transplant-associated cases of viral CNS infection caused by WNV, rabies virus, lymphocytic choriomeningitis–like viruses and human herpes virus-6 (HHV6) are becoming more commonplace (Wilson, 2013). JC virus (JCV) is associated with the occurrence of progressive multifocal leukoencephalopathy (PML) and is also associated with immunomodulatory therapies but has a high occurrence in HIV/AIDS patients. The aim of this chapter is to give an overview of the major virus infections of the human CNS, focussing on the related neuroimmunology. A summary table is included (Table 7.1) to guide the reader through the virus groups and summarize the related diseases. For particular virus groups, extensive studies have elucidated underlying mechanisms of brain infection and the associated immune response, while information for other viruses is lacking. The information in this chapter reflects this balance. Table 7.1 Viruses, prions and neurological diseases in humans Human herpes viruses (HHVs; Table 7.1) cause a range of acute, subacute and long-term neurological diseases in both immunocompetent and immunocompromised individuals. This is manifested most commonly as encephalitis, but myelitis is also associated with varicella zoster virus (VZV), human cytomegalovirus (HCMV), Epstein–Barr virus (EBV) and human herpes simplex virus-2 (HSV2). EBV can also cause CNS lymphomas and VZV leukoencephalitis in immunocompromised patients (Kleinschmidt-DeMasters and Gilden, 2001). Herpes simplex virus-1 (HSV1) is the most common cause of cold sores (herpes labialis) and herpes simplex-2 (HSV2) of genital herpes. Herpes simplex encephalitis (HSE) is the commonest fatal sporadic encephalitis in humans in Western countries (Rozenberg, 2013), with HSV1 being the primary cause of HSE with an estimated 2–4 cases per one million individuals in Western countries (Conrady et al., 2010). However, there is evidence from cerebrospinal fluid (CSF) testing that HSV2 infection in the CNS is becoming more common, particularly in younger age groups (Peter and Sevall, 2001). Other less common presentations such as a symmetrical brainstem inflammation with lesions associated with vasogenic oedema also occur (Miura et al., 2009). In neonates and young children, disease presentation is more frequently diffuse and/or multifocal (Kleinschmid-DeMasters and Gilden, 2001). Genetic factors involved in HSE have been examined in susceptible DA and resistant PVG rats to infection. The calcitonin receptor protein was identified as potentially critical for infection and viral spread to the CNS and subsequent HSE development (Abdelmagid et al., 2012). In addition, HSV1 CNS infection in the context of an Apo E4 phenotype has been linked to Alzheimer’s disease (Dobson and Itzhaki, 1999; Wozniak et al., 2009). Following acute infection, HSV establishes latency in sensory ganglion cells (Bertke et al., 2009) where it is thought to remain dormant until reactivation. However, it has been demonstrated that both HSV1 and HSV2 are shed intermittently in asymptomatic as well as symptomatic individuals in saliva (Scott et al., 1997; Tronstein et al., 2011). Furthermore, it has been found experimentally that HSV1 causes localized incomplete or low-level lytic infection causing a persistent immune response during latency (Halford et al., 1996; Feldman et al., 2002). Daily treatment with the antiviral drug acyclovir significantly reduces the localized immune response within the ganglion during latency (Halford et al., 1997). Therefore, the virus continuously undergoes incomplete or low-level reactivation until such circumstances allow it to fully reactivate. Furthermore, Woszniak et al. (2009) have data (in situ PCR; Figure 7.1) which has led them to propose that the virus can also be reactivated in the CNS, in a similar manner to the peripheral nervous system, by events such as stress and general infections, and that repeated reactivation causes cumulative damage, which in individuals with a genetic predisposition can lead to Alzheimer’s disease (AD). They also suggest that acyclovir should be used in clinical trials in the early stages of AD (Itzhaki and Wozniak, 2012). Figure 7.1 Herpes simplex virus type-1 (HSV1) DNA is localized in senile plaques of Alzheimer’s disease brain. In situ polymerase chain reaction (PCR) was used to locate the HSV1 DNA in sections of human brain. In this method, which is far more sensitive than in situ hybridization, a digoxigenin-labelled probe is hybridized to the target sequence in the tissue section after PCR amplification; it thus allows simultaneous amplification and localization. To locate plaques, thioflavin S staining was used. Sections were viewed under fluorescent light to locate the plaques (green staining) and then under white light to reveal the HSV1 DNA (brown staining). (Wosniak and Itzhaki, unpublished data.) VZV is usually acquired in childhood when it causes varicella (chickenpox), following which the virus establishes a latent state in trigeminal and dorsal root ganglia which lasts throughout life. VZV subsequently reactivates, spontaneously or after specific triggering factors, including immunosuppression, to cause herpes zoster (shingles), which may be complicated by post-herpetic neuralgia and several other neurological complications including vasculopathy (Gershon et al., 2010; Kennedy and Cohrs, 2010). Congenital cytomegalovirus (CMV) infection is the leading infectious cause of mental retardation and hearing loss due to infection in utero in the developed world; the virus also causes CNS complications in immunocompromised individuals (Tsutsui et al., 2005; Cheeran et al., 2009). CMV is also the commonest infectious agent to affect allograft recipients. The virus can be reactivated in immunosuppressed individuals such as patients with AIDS, transplant patients, elderly people and individuals admitted to intensive-care units (Griffiths, 2004). In mouse models, it has been shown that neural stem and progenitor cells are the most susceptible to CMV infection in the developing brain. During brain development, lytic infection tends to occur in immature glial cells. In longer term infection, the virus preferentially infects neuronal cells and in developing brains it may become latent in neural immature cells. It has also been shown that brain disorders may occur long after infection by reactivation of the latent infection (Tsutsui et al., 2005). EBV infects approximately 95% of the human population and persists latently in the memory B cell pool throughout life. EBV is the primary cause of infectious mononucleosis. The virus is also associated with tumours, such as Burkitt’s lymphoma, nasopharyngeal carcinoma and gastric cancers (Kaneda et al., 2012). Neurological complications include meningoencephalitis and myelitis (Kleinschmidt-DeMasters and Gilden, 2001). It has been reported that a history of infectious mononucleosis approximately doubles an individual’s risk of developing multiple sclerosis (MS) (Ascherio et al., 2001; Lünemann and Münz, 2009) (see Chapter 9). However, whether EBV or other viruses are causal agents of MS is a long-standing but inconclusive area of research. In common with other viruses which have been implicated, disease mechanisms for EBV are hypothetical. These include (i) autoreactive T cells that could be activated by EBV through molecular mimicry, (ii) enhanced breakdown and presentation of self-antigens, (iii) expression of viral superantigens or (iv) bystander activation. Most contentious is a direct role for EBV infection in CNS inflammation as a consequence of potential selective enrichment of EBV-infected B cells in ectopic meningeal follicles and perivascular CNS inflammatory infiltrates in MS brain (Serafini et al., 2007; Peferoen et al., 2010; Owens et al., 2011; Owens and Bennett, 2012). HHV6 infection is usually asymptomatic and commonly occurs during childhood. Clinically the virus causes exanthema subitum, also known as roseala infantum or the ‘6th disease’ (Ward, 2005). However, HHV6 reactivation (as for CMV) can lead to serious systemic diseases, especially encephalopathy, that may be fatal in immune-compromised or grafted patients (Michael and Solomon, 2012). HHV6 transplacental infection can also lead to neurological complications in the newborn (Hall et al., 2004). HHV6 DNA was detected in a total of 52 out of 126 (41.3%) nasal mucous samples from autopsy specimens from MS and cancer patients, showing that the nasal cavity is a reservoir for HHV6. Furthermore, specialized olfactory ensheathing glial cells were demonstrated to support HHV6 replication in vitro. These results indicate HHV6 utilization of the olfactory pathway as a route of entry into the CNS (Harberts et al., 2011). Evidence for the role of different aspects of the immune response in the CNS in herpes virus infections comes from clinical studies and in vivo or in vitro experimental models. Natural killer (NK) cell knockout mice or NK cell–depleted mice are susceptible to HSV-induced lesions in the CNS (Nandakumar et al., 2008). Chemokines regulate leukocyte trafficking to inflamed tissues and play a crucial role in orchestrating the immune response to HSV1 infection. Control of viral infections is associated with production of CXCR3 and CCR5 agonists and recruitment of leukocytes bearing these receptors. However, single knockouts of CXCR3 and CCR5 exhibit a mild phenotype when compared to ablation of leukocyte subsets associated with these receptors, suggesting functional redundancy between these two and most likely other chemokine receptors (Wuest and Carr, 2008). Intercellular adhesion molecule-5 (ICAM5), an immune modulator in the CNS, interacts with neurovirulence factor UOL as higher numbers of lymphocytes, but unaltered soluble ICAM5 and chemokine levels, were detected in ΔUOL HSV1-infected mouse brains. This suggests that ICAM5 plays a critical role in modulating chemokine production in the CNS (Tse et al., 2009). Humans and mice lacking signal transducer and activator of transcription-1 (STAT1) display increased susceptibility to HSV CNS infection (Pasieka et al., 2011), while STAT4 has been shown to regulate antiviral interferon gamma (IFNγ) responses and disease severity during chronic HSV2 infections in humans (Svensson et al., 2012). IFN lambda (IFNλ), a newly identified member of the IFN family (Kotenko et al., 2003), has also been shown to have the ability to inhibit HSV1 infection of primary human astrocytes and neurons. Furthermore, IFNλ treatment of astrocytes and neurons inhibits or reduces suppressor of cytokine signalling-1 (SOCS1), a key negative regulator of the IFN pathway (Li et al., 2011), which leads to increased IFN production. Viral immune evasion has also been extensively studied with HSV. HSV1 glycoprotein E (gE) mediates cell-to-cell spread and functions as an immunoglobulin (IgG) Fc receptor (FcγR) that blocks the Fc domain of antibody targeting the virus or infected cell thus blocking both IgG Fc-mediated complement activation and antibody-dependent cellular cytotoxicity against the virus (Lubinski et al., 2011) Therefore, gE promotes immune evasion from IgG Fc-mediated activities and probably contributes to virulence when antiviral antibody is present, such as during recurrent infections. HSV1 has been found to decrease immune cell types by causing apoptosis. The virus induces de novo expression of Fas L on the cell surface and causes the death of interacting human CD4+ T cells, CD8+ T cells and NK cells (Iannello et al., 2011). Immune suppression functions of specific virus proteins have also been identified. The HSV1-encoded glycoprotein B (gB) manipulates the MHC class II processing pathway by perturbing endosomal sorting and trafficking of HLA-DR molecules (Temme et al., 2010). Furthermore, the viral proteins gC and gE have been shown to provide a shield against neutralizing antibodies that interfere with the interaction of the virus glycoproteins gB, gD and gH–gL. Fusion of the virus envelope with the cell plasma membrane, necessary for virus entry, is dependent on interaction of these four viral glycoproteins. The HSV1 latency-associated transcript (LAT) also appears to play a role in immune evasion. Using LAT(+) and LAT(−) infected mice, results were obtained which suggest a novel immune evasion mechanism whereby the HSV1 LAT increases the number of HSV1 antigen–positive dendritic cells (DCs) in latently infected trigeminal ganglia, and interferes with DC phenotypic and functional maturation (Chentoufi et al., 2012). HCMV may persist by evasion of immune reactions which promote virus clearance. The viral glycoprotein UL141 targets the tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) death receptors preventing apoptosis of infected cells (Smith et al., 2013). Viral immune evasion of antigen presentation by both MHC class I and II molecules occurs; by restricting the presentation of viral antigens during both productive and latent infection, HCMV limits elimination by the human immune system (Noriega et al., 2012; Hesse et al., 2013). HCMV encodes homologues of cellular cytokines and chemokines and their receptors, which facilitates viral evasion of immune clearance (McSharry et al., 2012). The EBV genes, BCRF1 and BNLF2a, encode the viral homologue of interleukin-10 (IL10) (vIL10) and an inhibitor of the transporter associated with antigen processing (TAP), respectively. Both of these factors are known immune-evasins in the lytic phase of EBV infection. vIL10 impairs NK cell–mediated killing of infected B cells, interferes with CD4+ T cell activity and modulates cytokine responses. BNLF2a reduces antigen presentation and recognition of newly infected cells by EBV-specific CD8+ T cells. Together, both factors significantly diminish the immunogenicity of EBV-infected cells during the initial, early phase of infection before latency is established. This may also reduce recognition of infected B cells in the CNS (Jochum et al., 2012). The mononegavirales are an order of viruses with single-strand negative-sense non-segmented RNA genomes (Table 7.1). Genera predominately associated with CNS infection are the morbilliviruses and the more newly recognized henipaviruses, within the family Paramyxoviridae. Mumps virus in the genus Rubulavirus and also within the Paramyxoviridae is primarily associated with meningitis rather than encephalitis. Lyssaviruses within the family Rhabdoviridae including rabies viruses are still a major cause of CNS infection in many countries. Bunyaviridae have negative-strand segmented genomes and include the newly recognized Toscana virus within the genus Phlebovirus, and are vector borne. The exception to this mode of infection are the hantaviruses which are spread by rodent bite but can also cause CNS involvement. One member of the hantaviruses, Puumala virus, is known to be associated with meningitis, encephalitis, mononeuritis and polyneuritis, including Guillain–Barré syndrome (Clement et al., 2000). Measles virus (MV), the only human pathogen within the morbilliviruses and one of the most infectious human viruses, remains an important cause of child morbidity and mortality worldwide, with the greatest burden in the youngest children. Most acute measles deaths are due to secondary infections that result from measles-induced suppression of immune responses (Schneider-Schaulies and ter Meulen, 2002).Neurological complications with MV are much less frequent than with particular veterinary members of the genus, namely, canine distemper virus, phocine distemper virus and cetacean morbillivirus. However, MV causes an acute post-infection encephalitis in 1 in 1000 cases following primary infection. This is suggested to be due to an autoimmune response, as virus entry to the brain is not thought to occur. In addition, MV causes two fatal infections of the CNS, the long-term MV complication subacute sclerosing panencephalitis (SSPE) and measles inclusion body encephalitis (MIBE) (Cosby et al., 2002). The latter occurs in immunocompromised individuals, including children who are HIV-positive (McQuaid et al., 1998). Approximately 1 in 10,000 children (with a higher incidence in males) develop SSPE (Takasu et al., 2003; Bellini et al., 2005). This long-term persistent infection is most common in individuals who contract their initial measles infection before 2 years of age (Norrby and Oxman, 1990). MV antigen and RNA is present in neurons, in oligodendrocytes and in a much lower percentage of astrocytes in SSPE brain (Figure 7.2). Viral RNA and antigens are also present in inflammatory cells in perivascular cuffs in the infected brain (Cosby et al., 2002). In the establishment of both SSPE and MIBE, the infection of brain endothelial cells (BECs) at the blood–brain barrier (BBB) is likely to play a major role in virus spread into surrounding brain tissue. A small number of BECs in some blood vessels in SSPE brain were shown to be infected (Kirk et al., 1991) and it is also possible that the virus infects these cells in cases of measles but rarely enters the CNS unless individuals are immunocompromised. If the virus does cross the BBB and enters the neuronal network, spread has been shown to be trans-synaptic. Exit from the cell by the budding and production of mature virus is not necessary, and the envelope proteins are not required (McQuaid et al., 1998). This lack of selective pressure allows the accumulation of mutations in genes encoding these proteins, which further contributes to the persistent state (Baczko et al., 1993). Figure 7.2 MV antigen visualized by confocal microscopy in the frontal lobe grey matter in SSPE brain. Virus is present in neurons, oligodendrocytes and extensive cellular processes. Inflammatory cells are seen surrounding virus infected cells. Virus (green); nuclei (red). (McQuaid and Cosby, unpublished data.) A number of studies have examined cytokine and chemokines in SSPE brain or animal model systems. IFNγ and TNF alpha (TNFα) were found to be expressed in endothelial and glial cells (Anlar et al., 2001). Furthermore, MV has recently been shown to up-regulate the virus receptor polio-like receptor-4 (PVRL4, also known as nectin-4) in human brain endothelial cells and cause apoptosis by induction of TRAIL (Abdullah et al., 2013). The inflammatory lesions in SSPE consist of various cell subtypes and cytokines localized in particular regions of the brain, and these show certain associations with clinical course. Using in situ hybridization, IL2, IL6, TNF and leukaemia inhibitory factor (LIF) mRNA have been demonstrated in cells in the inflammatory infiltrate as well as in glial cells in foci of infection in cases of SSPE. LIF mRNA was also present in neurons in these areas. In the same study, LIF was also produced after MV infection in a human neuronal cell line (McQuaid et al., 1997). In contrast, the type-I IFN inducible human MxA protein, which exhibits antiviral activity against a number of RNA viruses, including MV, was reported to be highly expressed in the area surrounding, but not in the centre of, MV-antigen-positive lesions in SSPE brains. This pattern of MxA expression indicates that newly infected cells release type I IFN and are demarcated by a protecting barrier of MxA-expressing cells. Expression of MxA was found mainly in the cytoplasm of astrocytes and did not correlate with the presence of infiltrates of inflammatory cells, although some lymphoid cells were positive for MxA (Ogata et al., 2004). Proinflammatory chemokines were found to be produced in a transgenic mouse model of neuron-restricted MV infection as well as in neurons derived from these mice. The chemokines were IFNγ inducible protein (IP10), C–X–C motif chemokine-10 (CXCL10), monokine inducible by IFNγ (Mig), CXCL9 and RANTES (CCL5) (Patterson et al., 2003). Overall, the above studies indicate that neurons as well as glia play important roles in the protective antiviral response to MV in the CNS. The pattern of inflammatory cell infiltration has been studied in the frontal brain biopsies of 28 cases of SSPE by immunohistochemistry. Lymphocytic infiltration and gliosis were common pathologic findings. CD4+ T cells were observed in perivascular areas, and CD8+ lymphocytes in the brain parenchyma. B cells were located in large perivascular cuffs associated with a longer and slower disease course (Anlar et al., 2004). The T cell response in SSPE was also investigated by examining proliferation and cytokine secretion of cells from 35 patients and 42 healthy controls in response to virus and a number of CNS antigens. Proliferation in response to myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG) and the heat shock protein alpha B-crystallin (HSPB5) did not differ between the groups. IL12 secretion of peripheral blood leukocytes from SSPE patients to MV antigen was lower than in controls, and IFNγ, IL12 and IL10 production was also impaired in SSPE patients. The results did not demonstrate any bystander cellular response against myelin antigens, indicating that the CNS is not a predominant target of an autoimmune response in SSPE (Yentür et al., 2005) unlike post-infection encephalitis. A number of studies have been carried out to investigate MV clearance from the CNS. Investigation of humans with genetic or acquired deficiencies of either the humoral or cellular arm of the immune system, and rodent models, have implicated T cells in the control of ongoing MV infection. Using a transgenic mouse model in conjunction with depletion and reconstitution of individual B and T cell subsets alone or in combination, it was shown that neither CD4+–CD8+ T cells nor B cells alone are responsible for virus clearance. However, combinations of either CD4+ T cells or B cells, or of CD4+ and CD8+ T cells, are essential but CD8+ T cells with B cells are ineffective in control of virus levels. IFNγ and neutralizing antibodies to MV, but not perforin or TNFα alone, are associated with clearance of infection. TNFα combined with IFNγ is more effective in protection than IFNγ alone (Tishon et al., 2006). A more recent study has shown that primed T cells directly act to clear MV infection in the CNS by using a non-cytolytic IL12 and IFNγ-dependent mechanism and that this mechanism relies upon Janus kinase (Jak)–STAT signalling (Stubblefield-Park et al., 2011). Therefore, lack of an appropriate IFNγ response is likely to be a major factor in establishment of persistence of MV in the CNS. Current treatment for SSPE may slow the disease to some extent and comprises IFNα, ribavirin and inosiplex, either separately or in combination (Solomon et al., 2002). However, clearance when the virus is in a long-term persistent state is unlikely to occur. The role of regulatory T cells (Tregs) as regulators of the immune response in the brain in MV infection has recently been investigated (Reuter et al., 2012). It was found that persistent CNS infection in mice can be modulated by manipulation of Tregs in the periphery. CD4+ CD25+ Foxp3+ Tregs were expanded or depleted during the persistent phase of the CNS infection, and the virus-specific immune response and level of virus replication were analysed. Expansion of Tregs increased virus replication and spread in the brain. In contrast, depletion of Tregs induced an increase of virus-specific CD8+ effector T cells in the CNS and caused a reduction of infection. Nipah (NiV) and Hendra (HeV) viruses are the two members of the genus Henipavirus and are highly pathogenic, causing fatal encephalitis and respiratory disease in humans. Since the initial outbreak of HeV in Australia (1994) and NiV in Malaysia (1998), they have continued to cause sporadic outbreaks resulting in fatal disease (Mahalingam et al., 2012; Rota and Lo, 2012). The pathological features in human acute henipavirus infections comprise vasculopathy (vasculitis, endothelial multinucleated syncytia and thrombosis), microinfarcts and parenchymal cell infection in the CNS. This pathology also occurs in the lung, kidney and other major organs. Viral inclusions, antigens, nucleocapsids and RNA are readily demonstrated in the blood vessel wall and in numerous types of parenchymal cells. In addition to acute disease, relapsing henipavirus encephalitis is a rare complication reported in less than 10% of survivors. Pathological evidence suggests that this is due to viral reactivation in the CNS (Wong and Tan, 2012). The Bunyaviridae family contains more than 258 viruses and is divided into five genera, four of which comprise animal viruses some of which present serious risks to humans. The family includes Phleboviruses (e.g. Rift Valley fever (RVFV)), Nairovirus (e.g. Crimean–Congo haemorrhagic fever (CHFV)), Bunyavirus and Hantaviruses including some of the haemorrhagic fevers (Charrel et al., 2004). Toscana virus is a newly emerging virus transmitted by phlebotomus sandflies and initially discovered in central Italy, but it has also now been found in other European countries. Hantaviruses (e.g. Hantaan virus) are another, emerging group of viruses causing epidemics in the United States. Bunyavirus is the largest genus (Calisher, 1994), and symptoms caused by members include fever, rash, arthritis, encephalitis and hepatitis. These infections are common in Africa and South America. In the United States, only the California group of viruses are present, one of which is the La Crosse virus. Clinical features of this infection range from a mild febrile illness to aseptic meningitis and fatal encephalitis. Most children with symptomatic disease have signs of encephalitis, with nearly one-half having seizures (Rust et al., 1999). Viruses in the Lyssavirus genus in the family Rhaboviridae have a distinctive bullet-like structure. Rabies (Latin meaning ‘mad’) virus (RABV) is the type species of the genus Lyssavirus
CNS Infections
Introduction
Genome
Family
Genus or subgroup
Viruses
Neurological diseases
DNA, double stand
Herpesviridae
Alpha
HSV1/2, VZV
Sporadic encephalitis, myelitis latency in sensory ganglia, acute meningitis
Beta
HHV6, HCMV
Encephalitis in immunocompromised, neurological complication in newborn
Gamma
EBV
meningoencephalitis and myelitis
Papovaviridae
Polyoma virus
JC
PML
RNA, single strand, negative sense
Paramyxoviridae
Morbillivirus
Measles
Post infection encephalitis, SSPE, MIBE
Rubulavirus
Mumps
Meningitis
Henipaviruses
Hendra, Nipah
Encephalitis with vasculitis
Bunyaviridae
Bunyavirus
150 viruses transmitted by mosquitoes, including California group (e.g. La Crosse virus)
Encephalitis, meningitis
Phlebovirus
50 viruses transmitted by mosquitoes or phlebotomine flies (e.g. RVFV and Toscana)
Encephalitis
Nairovirus
CCHV
Encephalitis
Hantavirus
Transmitted by rodents (e.g. Hantaan and Puumala)
Encephalitis, meningitis, mononeuritis and polyneuritis, including Guillain–Barré syndrome
Rhaboviridae
Lyssavirus
Rabies and bat lyssaviruses transmitted by animal bite
Encephalitis (furious and dumb forms)
RNA, single strand, positive sense
Togaviridae
Alphavirus
WEEV, VEEV, EEEV, WEV
Encephalitis
Rubivirus
Rubella
Congenital defects, rare post-infection encephalitis
Flaviviridae
Flavivirus
E.g. WNV, TBEV, JEV
Encephalitis
Picornaviridae
Enterovirus
Poliovirus
Myelitis
Enterovirus
CV, EV71
Encephalitis, meningitis, associated delayed neuropathologies
RNA with DNA intermediate
Retroviridae
HIV
Dementia, neuroAIDS, CNS infection due to numerous opportunistic infections
HTLV1
Myelopathy, tropical spastic paraparesis
Infectious protein
Prions
Human diseases
CJD, vCJD, GSS, FFI, kuru
Herpes viruses
Herpes simplex viruses-1 and -2
Varicella zoster virus
Cytomegalovirus
Epstein–Barr virus
Human herpes virus-6 (HHV6)
Immune response in herpes virus infections
Immune evasion in herpes virus infections
Mononegavirales
Measles virus
Neuropathology
Cytokines and chemokines
T cell responses
Virus clearance
Henipaviruses
Bunyaviridae
Lyssaviruses
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel







Full access? Get Clinical Tree


