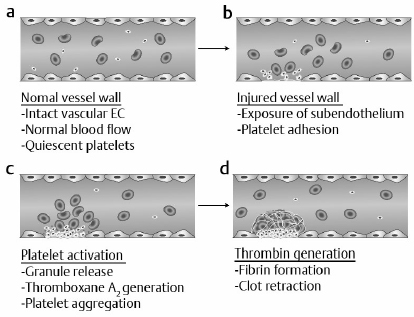
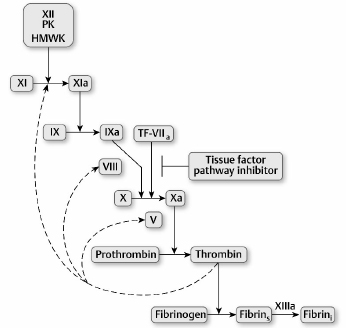
2 Hemostasis mechanisms prevent excessive blood loss after vascular injury while also preventing an inappropriate thrombotic response. This equilibrium is maintained by balanced interactions among vascular endothelium, platelets, and coagulation proteins that are present in blood plasma. Vascular trauma initiates primary hemostasis, which involves adhesion of platelets to damaged endothelium followed by platelet aggregation. The platelet thrombus is subsequently reinforced by a fibrin meshwork generated by the coagulation mechanism (secondary hemostasis). Vascular patency is restored by the fibrinolytic mechanism. Defects in either primary hemostasis (platelet number or function) or secondary hemostasis (coagulation factor deficiency) may lead to excessive bleeding.1 Vascular injury results in exposure of subendothelial components (e.g., collagen) that induce binding of von Willebrand factor (vWF) to platelets. The platelet vWF receptor, glycoprotein (GP)1b is required for this interaction. The subendothelium–vWF–GP1b mechanism leads to platelet adhesion. Subsequent platelet activation results in platelet aggregation, an event mediated by fibrinogen and platelet GPIIb/IIIa. Deficiency of GP1b, GPIIb/IIIa, vWF, or thrombocytopenia, or platelet dysfunction may result in defective primary hemostasis and bleeding.1 Reinforcement of the platelet thrombus requires initiation of the coagulation mechanism by tissue factor, a protein not normally exposed to blood. However, with vascular injury, tissue factor activity is expressed and initiates the process of thrombin generation. Blood coagulation involves a cascade of enzymatic conversions of an enzyme precursor (zymogen) to a protease (active enzyme), which subsequently activates another zymogen. This cascade ultimately leads to conversion of prothrombin to thrombin and, finally, to conversion of soluble plasma fibrinogen to an insoluble fibrin clot.2 Blood platelets are anucleate disk-shaped cells generated by bone marrow megakaryotes. With megakaryocyte maturation, the megakaryocyte cytoplasm demarcates into individual platelets that are shed into the circulation. The platelet membrane contains key GP receptors that mediate primary hemostasis, GP1b and GPIIb/IIIa. Platelets also contain granules that participate in primary hemostasis: α-granules containing proteins such as vWF and platelet factor 4, and δ-granules containing adenosine diphosphate (ADP), adenosine triphosphate (ATP), and serotonin. Release of serotonin from platelets mediates vasoconstriction following vascular injury.1 Following platelet adhesion to the damaged vessel wall, platelet activation occurs. Platelet activation is associated with granule secretion (platelet release reaction) and stimulation of prostaglandin synthesis. In the latter mechanism, arachidonic acid generated from platelet phospholipids is converted by platelet cyclooxygenase and thromboxane synthase to thromboxane A2, a potent platelet activator and vasoconstrictor. The granule release reaction and thromboxane A2 synthesis are also coordinated with expression of the GPIIb/IIIa receptor, fibrinogen binding to platelets, and platelet aggregation. These events result in a rapidly propagating platelet thrombus and cessation of bleeding.2 The activated platelet surface expresses receptors for coagulation proteases that ultimately generate fibrin to consolidate the platelet thrombus. Fig. 2.1 summarizes the hemostasis events following vascular injury. Although shown as discrete events, primary and secondary hemostasis occur nearly synchronously. Fig. 2.1a–d Summary of the hemostatic response to vascular injury. (a) In the normal state, vascular endothelium is intact, blood flow is normal, and platelets circulate in a nonactivated state. (EC, endothelial cells.) (b) With vascular injury, subendothelium components such as collagen are exposed, leading to platelet adhesion (mediated by von Willebrand factor [vWF] and platelet glycoprotein [GP]Ib). (c) With platelet activation, granule contents are released (adenosine diphosphate [ADP], serotonin), leading to vasoconstriction and platelet aggregation (mediated by fibrinogen and platelet GPIIb/IIIa). Generation of thromboxane A2 recruits additional platelets into the growing platelet thrombus. (d) Activation of platelets promotes initiation of coagulation in which thrombin generation leads to fibrin formation, reinforcement of the platelet thrombus, and subsequent clot retraction and consolidation. Activation of coagulation occurs by two pathways. The major pathway involves tissue factor expression that in the presence of factor VII (or activated factor VIIa) rapidly activates factor X to factor Xa, and factor IX to factor IXa. In the presence of factor V (Va), factor Xa converts prothrombin to thrombin. Thrombin cleaves fibrinogen to generate soluble fibrin, activates platelets, and also activates factors V, VIII, XI, and XIII. Soluble fibrinogen is cross-linked by factor XIIIa to generate the insoluble fibrin clot.2 These events are summarized in Fig. 2.2. Alternately, thrombin can be generated by the factor XII pathway (Fig. 2.2). An abnormal surface converts factor XII to XIIa. Factor XII converts prekallikrein to kallikrein, which amplifies factor XII activation, leading to activation of factor XI. These reactions require a cofactor protein—high molecular weight kininogen (HMWK). Factor XIa subsequently activates factor IX, and factor IXa in the presence of factor VIII (VIIIa) converts factor X to Xa.2 Although important for in vitro thrombin generation, the factor XII pathway is not important for in vivo thrombin formation because deficiencies of factor XII, prekallikrein, and HMWK are not associated with a bleeding disorder. This implies that there are in vivo pathways to initiate activation of factor XI, because factor XI deficiency does result in a bleeding disorder. Current data suggest that thrombin generated by the tissue factor pathway activates factor XI in a feedback loop in addition to factors V and VIII to amplify coagulation3 (Fig. 2.2). Fig. 2.2 The coagulation mechanism. Coagulation is initiated in vivo by expression of tissue factor (TF) activity. The TF–factor VII complex activates factors IX and X to IXa and Xa before inhibition of TF activity by tissue factor pathway inhibitor occurs. Thrombin generated by initial TF feeds back to activate factors V, VIII, and XI to amplify coagulation. Factor XII initiated-coagulation (with prekallikrein [PK] and high molecular weight kininogen [HMWK]) is important when artificial surfaces are present, but not for in vivo coagulation. Soluble fibrin generated by thrombin cleavage of fibrinogen is cross-linked by factor XIIIa to produce the insoluble fibrin clot. (Fibrins, soluble fibrin; Fibrini, insoluble fibrin.)
Coagulation and Platelet Plug Formation
Primary Hemostasis
Secondary Hemostasis
Platelet Physiology and Function
Coagulation Pathways
Related posts:
 Principles of Blood and Volume Replacement
Drugs Affecting Coagulation and Platelet Function
Principles of Blood and Volume Replacement
Drugs Affecting Coagulation and Platelet Function
 Intraoperative Non-Hematologic Adjuvant Methods for Preventing Blood Loss
Intraoperative Non-Hematologic Adjuvant Methods for Preventing Blood Loss
 Pediatric Neurosurgery–Specific Patient/Case Examples
Pediatric Neurosurgery–Specific Patient/Case Examples
 Neuroendovascular-Specific Patient/Case Examples
Neuroendovascular-Specific Patient/Case Examples
 Intraoperative Spine Surgery–Specific Patient/Case Examples
Intraoperative Spine Surgery–Specific Patient/Case Examples



