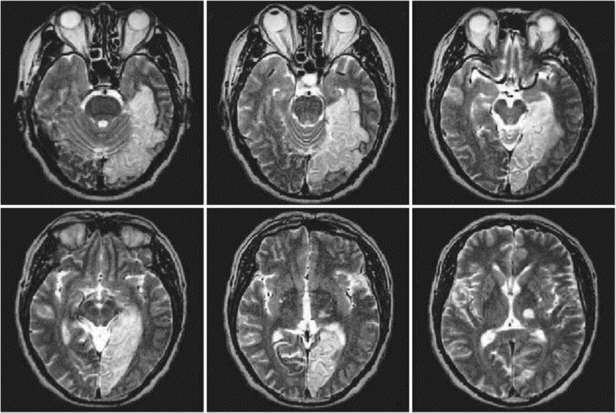
Magnetic resonance T-weighted images of the patient described in Box 19.1, showing an infarct in left temporal area (A: first stroke), the other infarct in right frontal (B: second stroke), and the third in right parietal area (C).
The total lesion volume size, the number of lesions, and the location of individual lesions are critical factors in the pathogenesis of MID (e.g., in general, the larger the infarct size, the more severe the dementia) [19, 20]. In contrast, quantitative correlations between the degree of cognitive deficits and lesion volumes or lesion locations have not been clearly elucidated [21]. The NINDS-AIREN criteria provide only a limited suggestion that bilateral anterior cerebral artery (ACA) distribution, posterior cerebral artery (PCA) distribution, parietotemporal and temporo-occipital association areas, and superior frontal and parietal watershed territories are considered the major candidate areas for VaD [14, 15, 16]. Multiple infarctions of any combination of these restricted cortical regions can result in MID.
In contrast to MID, even a single cortical infarction can lead to dementia. Damage to critical regions such as the angular gyrus or anterior cingulate gyrus also may cause cognitive deficits that lead to VaD [22, 23]. Therefore, the behavioral neurology of MID or single cortical infarct dementia requires understanding the cognitive and behavioral deficits following cerebral cortical infarction in distribution of ACA, PCA or middle cerebral artery (MCA), which we briefly review in this section.
Anterior cerebral artery territory infarction
The hemispheric branches of the ACAs supply the medial frontal surface (supplementary motor area; paracentral lobule, cingulate gyrus), the inferior frontal surface, and part of the medial parietal lobes (anterior portion of precuneus). The pericallosal branches of the ACAs supply the anterior four-fifths of the corpus callosum [24]. Therefore, ACA infarction mainly causes medial and inferior frontal dysfunction.
The functions of the frontal lobe are too broad and complex to detail here but include elementary motor function, praxis, speech/language output, attention, working memory, executive function, social judgment, and comportment [25]. Consequently, frontal lobe damage may result in a variety of behavioral and cognitive disorders, which can be classified under three major categories: first, executive dysfunction followed by dorsolateral frontal lobe damage; second, akinetic mutism or the apathy–abulia spectrum in medial frontal damage; and, third, disinhibition or aquired sociopathy in orbitofrontal damage ([26], Figure 19.2). Dorsolateral frontal lobe dysfunction will be discussed in the section on MCA infarctions.
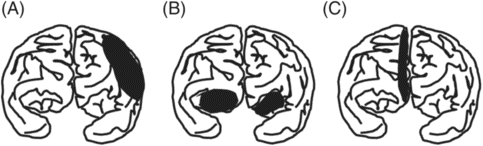
Frontal lobe dysfunctions. (A) Dorsolateral frontal: executive dysfunction; (B) orbitofrontal: disinhibition or aquired sociopathy; (C) medial frontal: reduced motivation and spontaneity (akinetic mutism or abulia).
Akinetic mutism, the sign of bilateral damage to the medial frontal lobe, is the most extreme form of a loss of spontaneity or initiative. Patients with akinetic mutism make no effort to communicate verbally or by gesture. Abulia, the minor form of akinetic mutism, can also follow medial frontal damage. The patient seems to be indifferent and less interested in the environment and people around him or her, has little spontaneous verbal output, and responds to questions very briefly, often with prolonged response latencies. Left to his or her own devices, the patient stays indoors, seated and immobile.
Orbitofrontal lobe damage can produce disinhibition and impulsive behaviors. Impulse control failures result in excessive sexual drives, voracious appetites, and addiction to alcohol, tobacco or drugs. Some patients exhibit compulsive behavior (e.g., cleaning, checking, arranging, ordering, hoarding or counting). One example would be a case reported by Hahm and his colleagues [27]. A 46-year-old patient showed a pathologic collecting behavior after a left orbitofrontal and caudate injury from an aneurysmal rupture of the anterior communicating artery (Figure 19.3A). Interestingly, his hoarding, an impulse control disorder or an ego-syntonic compulsion, was restricted to one specific item (toy bullets) (Figure 19.3B).
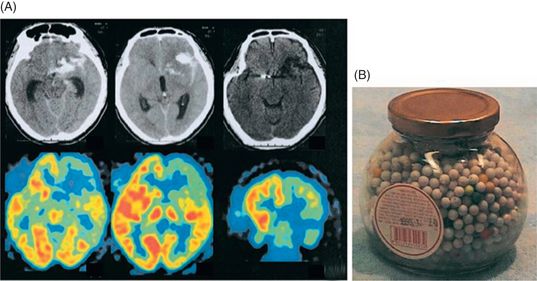
(A) Computed tomographic (CT) scans on admission (upper left two slices) show subarachnoid hemorrhage with a hematoma involving the left orbitofrontal region, which became a lowdensity lesion on a scan performed 2 years after onset (upper right slice). Fluorodeoxyglucose C [18F]-positron emission tomography performed 2 years after onset (lower row) shows glucose hypometabolism in the left caudate and frontal lobe, mildly in the anterior temporal area, and most prominent in the orbitofrontal region. (B) A sample bottle containing the toy bullets collected by the patient.
Comment
This patient meets NINDS-AIREN criteria for probable VaD since there were impairments of memory and in two or more cognitive domains, evidence of cerebrovascular disease by focal signs on neurologic examinations and brain imaging, and a temporal relationship between the dementia and the strokes. At the first attack, he had mild language impairment that improved to the extent that he was able to manage his daily activities. After the second stroke, however, abnormal behavior and impaired judgment led him to withdraw from complex activities, although this improved slightly over time. After the third stroke, he declined significantly and was no longer able to do basic activities such as dressing or eating. As we can see, there were clinical features of a stepwise deterioration over 2 years, with a fluctuating course associated with acute deficits followed by partial recovery. This was caused by three territorial infarctions without subcortical ischemic changes or lacunes and led to the diagnosis of MID.
Other behavioral disorders associated with orbitofrontal damage may include utilization and imitation behaviors. Utilization behavior has been described as “a disturbance in responses to external stimuli, so that a patient with utilization behavior simply takes and uses the object presented to them” [28]. Imitation behavior is characterized by “patients imitating the gestures and behavior of the examiner without having been asked to do so, and continuing to imitate after being asked to stop” [28]. These two “environmental dependency” syndromes are classified as a unilateral orbitofrontal symptom, without clear relation to the hemispheric dominance, and are interpreted as a release of the parietal approach behavior to visual and tactile stimulation from the outside world [28, 29].
Acquired sociopathy is another frequent consequence of orbitofrontal damage [30]. Orbitofrontal lobe, gyrus rectus, and anterior cingulate gyrus serve to integrate sensory association cortex information (occipital lobe, temporal lobe, and parietal lobe) with limbic cortical control of emotional responses. Normally, people can be affected by external stimuli that drive positive or negative emotions in order to make a proper decision, but the patient with orbitofrontal or anterior cingulate gyrus lesions may not be able to appropriately understand or respond to the external stimuli, such that he or she cannot achieve normal social or emotional decision-making [31].
Heilman and Watson [32] suggested that effective interaction with the environment required the presence of two critical motor systems, which they called the “how” system (praxis system) and the “when” system (intentional system). Disorders of the “how” or praxis program are called apraxias, while damage to the “when” or intentional programs are referred to as motor intentional disorder or action-intentional disorder. The pathophysiology for ideomotor apraxia is controversial. Damage to the anterior corpus callosum can result in left-hand apraxia because it disconnects the left hemisphere language areas from the right movement control area for the left hand (language motor disconnection; Wernicke 1874 [33]). Geschwind [34, 35] postulated that auditory stimuli from primary auditory cortex (Heschl’s gyrus) are conveyed to the auditory association cortex (Wernickes’s area) in the left hemisphere, which is connected to premotor cortex by the arcuate fasciculus; the premotor cortex on the left is connected to the left primary motor cortex. The information in the left premotor cortex can also be conveyed to the right premotor cortex through the anterior corpus callosum. According to Geschwind’s schema (Figure 19.4), any lesions of this pathway (premotor cortex, anterior corpus callosum, arcuate fasciculus) can cause ideomotor apraxia.
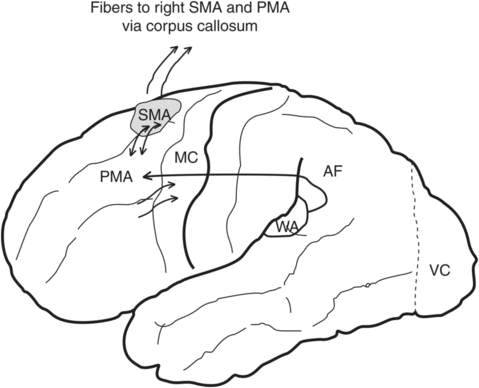
Motor intentional disorder is divided into four different types: [1] akinesia, a failure of initiation of movement in the absence of a corticospinal or motor neuron lesion; [2] hypokinesia, a delay in initiating a response; [3] motor impersistence, the inability to sustain a movement or posture; and [4] motor perseveration, the inability to stop a movement or an action program (Figure 19.5). While ideomotor apraxias are usually associated with left hemisphere dysfunction [215], right hemisphere damage may be dominant for intentional control of the motor systems [36]. Networks that mediate the intentional systems are widely distributed and have not been fully elucidated, but the fact that the frontal lobes play a critical role is supported by many human lesion studies and experimental animal studies: limb akinesia for medial frontal lesions [37], directional limb hypokinesia for frontoparietal lesions [38], motor impersistence for dorsolateral frontal lesions [39], and motor perseveration for frontal subcortical lesions [40].

Perseveration on the Ogden copying task by a 65-year-old man with acute left frontotemporal infarction.
Patients with infarctions of the anterior corpus callosum can display a callosal disconnection syndrome, including left-hand ideomotor apraxia, left-hand agraphia, left-hand tactile anomia, right-hand acopia, and intermanual conflict [41]. Recently, Seo et al. [42] reported that after an infarction involving the right medial frontal lobe and corpus callosum, a 66-year-old right-handed man demonstrated right limb motor impersistence on bedside evaluation, which was substantiated experimentally. This suggested that following a callosal lesion, motor impersistence occurs more frequently in the dominant than the non-dominant limb. Secondary mania featuring euphoria, pressured speech, and hyperactivity is frequently associated with lesions of orbitofrontal cortex, almost always in the right hemisphere [43].
The elementary neurologic symptoms and signs of ACA stroke are contralateral to motor and sensory deficits (leg predominant weakness). Bilateral ACA occlusions can produce paraparesis with or without sensory loss. Pathologic reflexes such as grasp, groping, snout, and sucking reflexes may appear unilaterally or bilaterally in hands, feet or around the mouth [44]. Urinary incontinence and disturbance of sphincter control have been described in superior frontal and cingulate gyrus damage [45].
Middle cerebral artery territory infarction
Cortical areas supplied by the MCA include, superiorly, the frontal, parietal, and occipital convexities and, inferiorly, the temporal convexity. Not surprisingly, cognitive and behavioral deficits resulting from MCA infarction are as variable as the location of the lesion.
As noted above, dorsolateral prefrontal cortex is a critical area for executive function (Figure 19.2). Executive functions include planning, goal monitoring, and fluency, and flexibility of thought in the generation of solutions. Patients with dorsolateral prefrontal damage may not be able to formulate a plan of action, consider possible different plans (impairment of mental fluency) or switch from one plan to another, which is required for success of ongoing actions (impairment of cognitive setshifting). Motor intentional disorders such as directional hypokinesia, motor impersistence, and motor perseveration have also been reported in dorsolateral prefrontal lesions, mostly when the right hemisphere is affected.
The typical clinical picture of superficial MCA territory infarctions includes sudden onset of contralateral sensorimotor deficits, with aphasia in left hemispheric (dominant hemisphere) lesions and visuospatial impairment and neglect syndrome in right hemispheric (non-dominant hemisphere) lesions. The relative severity of motor deficits from mild hemiparesis to complete hemiplegia depends on the location and size of the infarction. Hemisensory loss affecting all sensory modalities can be produced, as can cortical sensory loss with agraphesthesia, astereognosis, and failure of two-point discrimination. Approximately 90–95% of right-handed individuals have language dominance in the left hemisphere [46], so left MCA infarctions frequently cause aphasia syndromes. Rarely, right hemisphere lesions can produce aphasia in right handers [47]. Broca’s aphasia consists of non-fluent, effortful speech with relatively preserved comprehension and follows damage to the left inferior frontal gyrus (Broca’s area: pars opercularis and pars triangularis) and its adjacent areas. The hallmark of Wernicke’s aphasia is fluent speech with disturbance of auditory comprehension, derived from the damage of the posterior one-third of the superior temporal gyrus and its adjacent areas. Global aphasia is caused by a large stroke encompassing Broca’s and Wernicke’s areas (Figure 19.6). Patients with global aphasia have decreased spontaneous speech and comprehension and impaired repetition, reading, and writing [48, 49, 50]. Damage to the arcuate fasciculus, the fibers connecting the Broca’s and Wernicke’s areas, and the inferior parietal lobule (supramarginal gyrus) causes conduction aphasia characterized by impaired repetition and preserved comprehension [51]. Transcortical aphasias show preserved repetition. They result from the lesions affecting structures surrounding perisylvian language centers (Broca’s and Wernicke’s areas) with spared perisylvian cortex and arcuate fasciculus [52]. Destruction of the dominant supplementary motor area is a common pathogenic mechanism for transcortical motor aphasia [53] and lesions in the temporoparieto-occipital area for transcortical sensory aphasia [54].
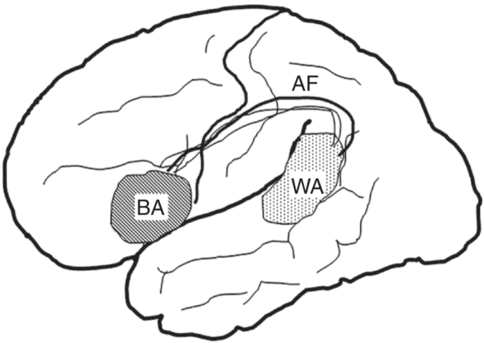
Perisylvian language centers and their connections. BA, Broca’s area; WA, Wernicke’s area; AF, arcuate fasciculus.
Ideomotor apraxia is defined by the inability to perform previously learned or skilled movements in response to commands that cannot be explained by weakness, sensory loss, abnormal movement, poor comprehension or inattention. A lesion at any point along Geschwind’s schema (see Figure 19.4) can cause an ideomotor apraxia. Alternatively, Heilman and colleagues [215] suggested that knowledge of motor skills, or the time–space–motor representation (praxicon), is stored in the dominant parietal cortex (angular gyrus, supramarginal gyrus). Therefore, left parietal lesions can also produce ideomotor apraxia.
The posterior part of inferior parietal lobule (angular gyrus) is a heteromodal association cortex that responds to stimulation in more than one sensory modality [55]. Damage to this higher-order, supramodal cortex gives rise to impairments of multimodal interaction related to praxis (see above) and language, such as anomia, alexia, and Gerstmann’s syndrome (acalculia, agraphia, finger agnosia, and right–left disorientation [56]), which together compose the angular gyrus syndrome.
Given that the right posterior parietal lobe is dominant for visuospatial integration and spatial attention [57, 58], lesions of the right parietal multimodal association cortex can yield visuospatial dysfunction and a neglect syndrome. Visuospatial function is categorized into visuoperceptual function, geographical orientation, and visuoconstructive ability [59]. Visuoperceptual function is considered as an ability to discriminate angles, shapes, and colors of the presented object, or decide whether two faces are the same or different. Geographical orientation refers to the selective way-finding ability within a three-dimensional environment or two-dimensional map. The ability to copy, draw, or construct two- or three-dimensional figures or shapes can be defined as visuoconstructive function. Though visuospatial dysfunction predominantly follows right-sided posterior lesions, visuoperceptual and visuoconstructional impairments can follow bilateral posterior injury [60].
Unilateral spatial neglect is a clinical syndrome in which patients are unaware of or fail to explore stimuli located in the contralesional half of extrapersonal space, even in the absence of primary sensory or motor deficit [61, 62, Figure 19.7]. It can result from a variety of lesions, both cortical and subcortical, but the temporoparietal cortex is known to be one of the most critical anatomical substrates for hemispatial neglect [63, 64, 65, 66, 67]. Patients with neglect syndrome often exhibit reluctant or reduced movement toward the contralesional space, even though they have no sensory attentional problems. This type of neglect is called motor-intentional neglect, as opposed to sensory-attentional neglect [62]. As mentioned above, since the right frontal cortex is essential for motor intention, lesions causing intentional neglect usually include the right frontal area [216]. Patients with neglect syndrome sometimes deny or fail to recognize their hemiplegia (anosognosia for hemiplegia; [68]) or that their contralesional extremities are their own (personal neglect or asomatognosia; [69]).
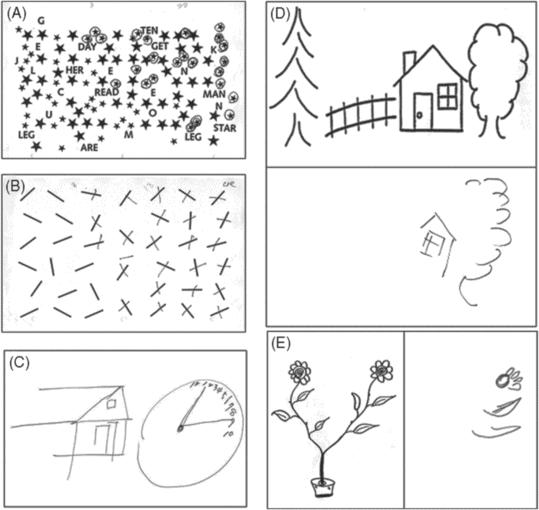
Examples of left hemispatial neglect. A; star cancellation task, B; modified version of Albert’s line cancellation task, C; spontaneous drawing (house, clock), D; copying modified Ogdenscene [217], E; copying two daisy figures.
In addition to visuospatial function and spatial attention, the right parietal lobe is dominant for affective prosody. Consequently, patients with right hemisphere injury have greater difficulty regulating and understanding emotional components of language than patients with left hemispheric lesions [70, 71].
In terms of psychiatric symptoms, delirium or an acute confusional state is most commonly associated with right MCA infarction involving the right middle temporal gyrus or inferior parietal lobule [72, 73]. In contrast, post-stroke depression can be the prominent manifestation of left MCA infarction affecting the left fronto-opercular region [74, 75].
Posterior cerebral artery territory infarction
The PCA arises from the terminal bifurcation of the basilar artery and has four main cortical branches: the anterior temporal, posterior temporal, parieto-occipital, and calcarine arteries, which supply the occipital lobes and the inferomedial portions of the temporal lobes. The most common neurological deficit in the PCA infarction is a contralateral visual field defect caused by the lesion of the primary visual cortex, the optic radiation or the lateral geniculate body [76]. Complex visual abnormalities, such as simple or formed visual hallucinations localized to the affected visual field or visual perseveration (palinopsia and illusory visual spread), are often present in the PCA infarction [77, 78, 79]. Alexia without agraphia is a classic symptom that results from a lesion in the left calcarine cortex and adjacent splenium of the corpus callosum [80]. It is usually accompanied by color anomia [81]. The symptomatology is interpreted as a disconnection of the visual input to the intact right visual cortex from the left language area (Figure 19.8A). If the lesion extends to the left angular gyrus, alexia with agraphia, Gerstmann’s syndrome (acalculia, agraphia, right–left confusion, finger agnosia), and ideomotor apraxia are often present. Patients with left PCA infarction may have transcortical sensory aphasia, although aphasia is not common in left PCA infarction [82].
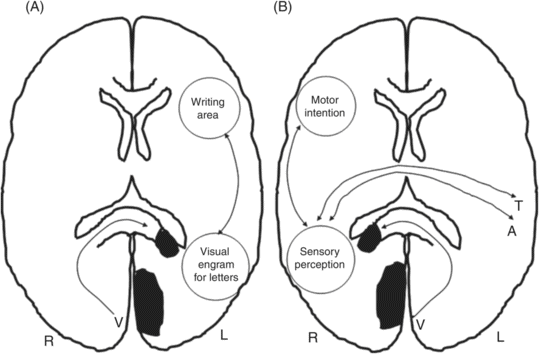
A. Alexia without agraphia. The visual information from the right visual cortex cannot reach the left inferior parietal language area because of a splenial lesion. B. Left spatial neglect limited to visual modality caused by right occipital and splenium lesion.
Luders et al. [83] observed that global aphasia was produced by electric stimulation of the dominant basal temporal region known as the basal temporal language area (BTLA). Interestingly, in the countries such as Korea or Japan where people use language that can be written in both ideogram (a graphic record of a meaning) and phonogram (a graphic record of a sound), dissociation between ideogram and phonogram impairment after brain injury has been reported. Kwon et al. [84] reported that a 64-year-old right-handed man, who used to be a Hanja (Korean ideogram) calligrapher, showed alexia with agraphia in Hanja (Korean ideogram) but preserved Hangul (Korean phonogram) reading and writing after a left posterior inferior temporal infarction (Figure 19.9), a similar area to the BTLA. This was consistent with previous reports about dissociation between Kanji (Japanese ideogram) and Kana (Japanese phonogram) processing [85, 86].
MRI scan of a patient with Hanja (Korean ideogram) alexia showing an infarct involving the left lingual, fusiform, and parahippocampal gyri and a lacune in the left thalamus.
The posterior temporal branch of the PCA supplies medial temporal structures (including the hippocampus), and unilateral occlusion of this artery (especially the left) causes infarction of the hippocampus and medial temporal lobe, which leads to temporary amnesia. However, bilateral occlusion can produce permanent amnesia [87]. Prosopagnosia, the inability to recognize familiar faces, is rarely developed by damage to the right fusiform gyrus (fusiform face area) [88]. If PCA infarction extends to the right parietal or temporal lobe, visuoconstructional disability and geographical disorientation can occur [89, 90]. Visuospatial neglect also can be elicited by a lesion combining the right occipital lobe and splenium, an analog lesion of the left occipital lobe and splenium injury associated with alexia without agraphia [91] (Figure 19.8B). This visual neglect might be related to a disconnection between the visual information processed by the left occipital lobe and the right posterior temporoinferior parietal areas that mediate attention in the left hemispace. Additionally, in a large number of studies regarding hemispatial neglect in PCA infarction, it was reconfirmed that only the right occipital plus splenial lesion significantly influenced the frequency and severity of neglect [92]. Bilateral destruction of primary visual cortex causes cortical blindness. Patients with cortical blindness are occasionally unaware of their visual loss. This is a phenomenon known as Anton’s syndrome, or visual anosognosia. The mechanism and precise anatomy of visual anosognosia remain uncertain. On the contrary, patients with cortical blindness might have some residual visual function (blindsight) but are unaware of it and deny its existence [93, 94].
Ungerleider and Mishkin [95] proposed that there are two parallel visual processing pathways: a dorsal or occipitoparietal “where” pathway for spatial perception and visuomotor performance and a ventral or occipitotemporal “what” pathway for object discrimination and recognition. Patients with bilateral occipitoparietal lesion have Balint’s syndrome, defined by a triad of simultanagnosia, the inability to recognize a picture or scene as a whole; optic ataxia, impaired hand movement under visual guidance; and gaze apraxia, an inability to direct gaze voluntarily toward the peripheral field [96]. Patients with bilateral occipitotemporal lesion have prosopagnosia, visual object agnosia (inability to identifying a visually presented object even with normal perception), and achromatopsia (acquired color blindness) [97, 98, 99].
Ischemic–hypoperfusive vascular dementia
Behavioral and neuropsychological findings in borderzone infarction
There are two types of borderzone infarction: the anterior borderzone infarct is located between the superficial territories of the ACA and MCA while the posterior type is the infarction located between the superficial territories of the MCA and PCA (Figure 19.10).

Fluid-attenuated inversion recovery (FLAIR) magnetic resonance images showing a right anterior borderzone infarction and left posterior borderzone infarction in a 92-year-old man who showed somnolence, abulia, and mild right hemiparesis. Neuropsychological tests performed 10 days after symptom onset showed frontal executive dysfunction and transcortical sensory aphasia.
The anterior type located in the frontal parasagittal borderzone area manifests as somnolence and transcortical motor aphasia [100, 101, 102, 103]. When the anterior borderzone infarct is located in the non-dominant hemisphere, mood disturbances such as apathy or euphoria may also develop [104]. Sometimes the anterior borderzones are affected bilaterally, which results in akinetic mutism and apathy as well as focal neurological deficit such as paraparesis mimicking spinal lesion, quadriplegia or triplegia, or bladder disturbance [100, 101, 102, 103].
The posterior type located in the parietotemporoccipital triangle produces Wernicke-type aphasia, hemispatial neglect, anosognosia, transcortical sensory aphasia, cortical hemihypesthesia, sensorimotor hemiparesis or hemianopia [100, 101, 102, 103].
Behavioral and neuropsychological findings in chronic hypoperfusion
The relation between chronic ischemia and cognitive functions in humans is not completely understood. However, a few reports suggested that cognitive impairments can be associated with the chronic cerebral hypoperfusion state caused by a variety of medical conditions [105, 106, 107]. For instance, cognitive impairment is common among elderly patients with systolic hypotension due to heart failure [107]. Reduced cerebral perfusion was associated with cognition, especially frontal and memory dysfunctions. Recently, cerebral blood flow velocity predicted worse performances in depression as well as attention and executive functions in patients with heart failure. COPD patients show cognitive decline of frontal type with the worsening of the hypoxemia [105]. It has been reported that the correction of a chronic cerebral hypoperfusion state can lead to recovery of mental decline [108]. Anemia is associated with visuospatial and frontal dysfunctions (Son et al., 2012) and depression in patients with mild cognitive impairments. Furthermore, anemia predicts an increased risk of developing dementia.
Cognitive impairments associated with chronic hypoperfusion seem to be non-specific, not as distinctive as those in anterior or posterior type of borderzone infarction [109]. They are characterized by slow onset and gradual progression. It has been known that periventricular white matter, basal ganglia [110], and hippocampus [111] are susceptible to chronic ischemic hypoperfusive states. Thus, interruption of prefrontal–basal ganglia circuits or hippocampal damage may explain the cognitive decline in these patients. Alternatively, reduced cerebral perfusion might affect cortical thinning in the frontal and temporal regions, which in turn leads to cognitive impairments.
Dementia associated with stroke of subcortical location
Behavioral and neuropsychological features in basal ganglia lesions
Caudate infarction
Patients with infarcts in the territory of the lateral lenticulostriate arteries show motor and neuropsychological deficits whereas those with infarcts in the territory of the anterior lenticulostriate arteries have relatively mild neuropsychological deficits [112].
Caudate infarction often results in abnormal behavior and cognitive impairment [112, 113, 114] (Figure 19.11). Hemichorea [115] can occur, but happens less often. Abnormal behaviors associated with caudate infarction include abulia, apathy, blunting of response and lack of initiative [112, 113, 114]. The mechanism of abulia can be explained by interruption of the limbic–frontal connection [114]. Agitation, anxiety, and talkativeness or disinhibition can also occur [114, 116]. Mendez et al. [113] reported that dorsolateral caudate involvement may cause decreased spontaneous verbal and motor activities and ventromedial lesions may result in disinhibited, inappropriate, and impulsive behavior. Rarely, patients with left caudate head infarction manifested recurrent delusional ideas without dementia [117].
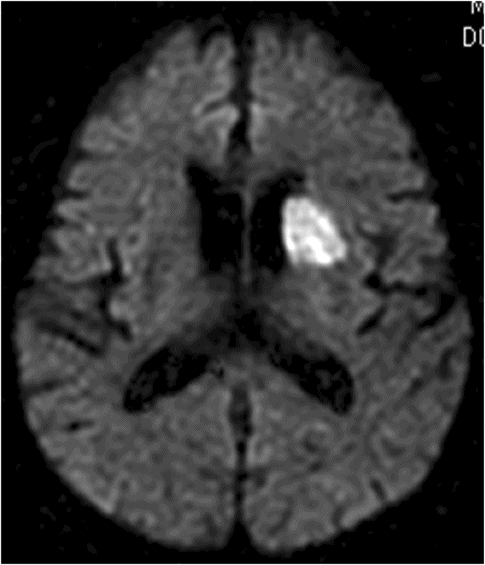
Diffusion-weighted magnetic resonance imaging showing a left caudate infarction in an 80-year-old man who presented with abulia, lack of spontaneity, and apathy. Neuropsychological tests, performed 10 days later, revealed frontal executive dysfunction, naming difficulty, and mild memory impairment with retrieval-defect type.
The common cognitive impairment is memory disturbance, with retrieval defect and aphasia [112, 113, 114]. Patients with left caudate lesions have verbal amnesia, while patients with right caudate lesions show visual amnesia [112]. Neuropsychological tests in caudate lesions show decreased free recall of episodic and semantic items, with good recognition memories scores [113]. These abnormalities have been explained by the disconnection of the caudate from the frontal lobe [112, 118]. Indeed, there was one study showing that patients with caudate infarction performed less well on memory, abstract reasoning, and frontal executive functions, and had significantly reduced metabolism in the prefrontal cortex [119]. A variety of aphasia such as transcortical motor aphasia, characterized by semantic and verbal paraphasias and perseverations without comprehension impairment, occurs in patients with a left caudate lesion [112, 113, 114, 120]. Alexander et al. [120] argued that acute disconnection of linguistic pathways between anterior and posterior speech areas, which are connected with the caudate nucleus, may yield aphasia.
Lentiform nucleus infarction
Unilateral lentiform nucleus infarction (putamen and globus pallidus) commonly causes movement disorders such as dystonia but rarely causes neurobehavioral disorders such as abulia or disinhibition [121]. It has also been reported that speech disturbance, obsessive–compulsive disorder, and auditory hallucinations may occur in these patients [122, 123]. Laplane et al. [123] considered that these psychiatric disorders in globus pallidal lesions could be related to disturbances in the circuit linking the frontal associative cortex and the basal ganglia. Rarely, patients with left globus pallidus infarction showed inattention, decreased verbal fluency, emotional blunting, and amnesia without any motor symptoms [124].
Capsular genu lesions
Cognitive impairment in capsular genu infarction (Figure 19.12) is characterized by fluctuating alertness, inattention, memory loss, apathy, abulia, and psychomotor retardation [125]. Several reports have shown that amnesia was the major presenting feature [126, 127, 128, 129] but other reports showed that pure abulia without other neurological deficits can occur [130]. The most prominent findings in capsular genu infarctions have been reported to be faciolingual and motor deficits as a result of the disruption of corticopontine and corticobulbar fibers [131], but other series have showed that pyramidal and corticobulbar tracts were minimally involved [125]. Essential features of cognitive impairment in capsular genu infarct are similar, with the clinical features found in polar [214] or paramedian thalamic infarction [218]. Rarely it was reported that obsessive–compulsive behaviors disappeared after a left capsular genu infarction [132].
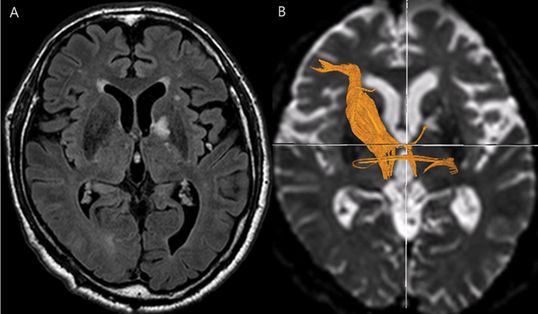
Fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging showing a left genu portion of the internal capsule including the globus pallidum in a 79-year-old man who presented with memory impairment and behavioral changes such as abulia and apathy. Neuropsychological tests, performed 30 days later, revealed frontal executive dysfunction, naming difficulty, and verbal memory impairment. (B) Diffusion tensor imaging shows an interruption of the white matter fibers in the left anterior thalamic radiation.
The mechanism for cognitive impairments associated with genu infarctions seems to involve interruption of the inferior [126] and anterior [133] thalamic peduncles. The ventral amygdalofugal pathway sends fibers to the dorsomedial nucleus of thalamus via inferior thalamic peduncles [134], and the efferent pathway of dorsomedial nucleus projects to the prefrontal cortex also through inferior thalamic peduncles [135]. Infarction of inferior thalamic peduncles that course in the vicinity of the genu of the internal capsule seemingly disconnects dorsal medial thalamus from amygdalae and the frontal cortex. Anterior thalamic peduncles convey reciprocal connections between the dorsomedial nucleus and the cingulate gyrus, as well as the prefrontal and orbitofrontal cortex [136]. Functional brain imaging showed a focal hypoperfusion in ipsilateral inferior and medial frontal cortex [130, 133] and hypometabolic activity in the temporal cortex ipsilateral to the capsular lesion [129]. Recently, we reported that patients with genu infarction had an interruption of the anterior thalamic radiation [137].
Behavioral and neuropsychological findings in thalamic lesions
Tuberothalamic arterial territory infarction
Cognitive and behavioral abnormalities after tuberothalamic artery infarction are anterograde amnesia, apathy, and frontal executive dysfunction (Figure 19.13). However, symptoms may differ depending on the hemisphere involved. Patients with left -sided lesions have transcortical aphasia, verbal and visual memory impairment, and acalculia; patients with right-sided lesions show hemispatial neglect, visual memory impairment, and disturbed visuospatial processing [214].
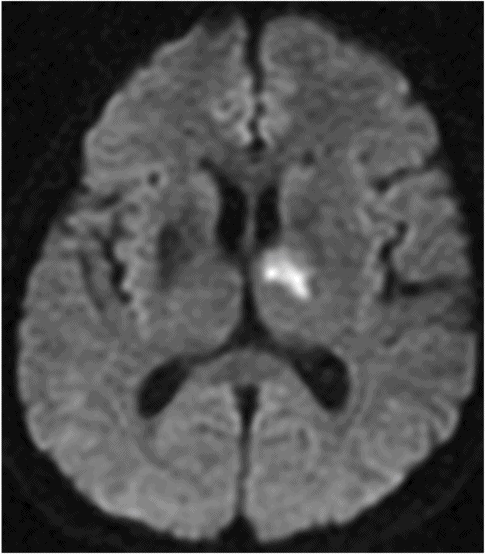
Diffusion-weighted magnetic resonance imaging shows a right anterior thalamic infarct involving the anterior nucleus and mamillothalamic fasciculus in a 63-year-old man who developed anterograde amnesia and abulia.
Memory disturbance associated with thalamic infarction is usually described as “diencephalic amnesia” [138, 139]. The pattern of memory loss in patients with a thalamic lesion resembles that seen after lesions in the medial temporal area [140]. The neuropsychological findings in patients with thalamic lesions are characterized by deficits of free recall and recognition but some studies suggest that the memory impairment is a retrieval deficit, that is, recognition better than recall [141, 142]. Another feature that has been described in amnesia related to tuberothalamic infarction is “palipsychism.” Ghika-Schmid and Bogousslavsky [141] reported that most patients with tuberothalamic infarction had palipsychism, which is the superimposition of temporally unrelated information during cognitive activities, with ongoing parallel simultaneous processing in more than one domain. It has also been reported that bizarre confabulations can occur, which are similar to those found after medial frontal lobe lesions [143]. These language disturbances might be explained by the thalamo-anterior temporal disconnection [147]. Rarely, aggressiveness in a patient was reported to disappear following an anterior thalamic infarction [148].
The anatomical basis of diencephalic amnesia remains unclear, but it has been reported that hippocampal-related neural structures such as the mamillothalamic tract and anterior thalamic nuclei are critical to memory function [139]. Many reports have emphasized that the mamillothalamic tract is responsible for anterograde amnesia. According to van der Werf et al. [149], the mamillothalamic tract was affected in 24 of 25 patients with diencephalic amnestic syndrome, whereas 11 of 13 patients with no or mild memory impairments despite thalamic lesions had an intact mamillothalamic tract. They argued that lesioning of the mamillothalamic tract is the best predictor of the occurrence of an amnestic syndrome.
As noted above, patients with thalamic lesions but intact mamillothalamic tracts usually have no amnesia or, if present, show mild amnesia. In these patients, frontal cortical dysfunction may explain the nature of the memory disorder, which shows evidence of frontal-type memory problems including impaired spatial working memory, increased forgetting rates, poor prospective memory, and inadequate elaborative encoding as well as frontal disinhibition [150]. Other than amnesia, neurobehavioral symptoms associated with tuberothalamic infarctions are apathy, abulia, perseveration, and, less often, disinhibition [141, 149, 151]. Ghika-Schmid and Bogousslavsky [141] stressed the importance of severe perseverative behavior, which is apparent in thinking, spontaneous speech, memory, and executive tasks.
Other abnormalities associated with tuberothalamic artery infarction include aphasia, especially transcortical motor aphasia; fantastic paraphasia; neologism [152]; neglect and topographic disorientation, especially after right-sided lesions [214]; hypophonia; dysarthria; and ipsilateral ptosis [141, 153].
Paramedian artery territory infarction
The essential features of an infarction in the territory of the paramedian artery (Figure 19.14) are clinical evidence of arousal disturbance, memory impairment, and vertical gaze palsy combined with impairment in attention span, orientation, intellect, and visual perception [154, 155, 156, 157, 158, 214]. Bilateral paramedian thalamic infarction can occur, since both thalamic regions are occasionally supplied from a common trunk on one side [155]. Outcome is better in right-sided than left-sided or bilateral infarcts. The remained cognitive deficits were related to frontal dysfunction [159].
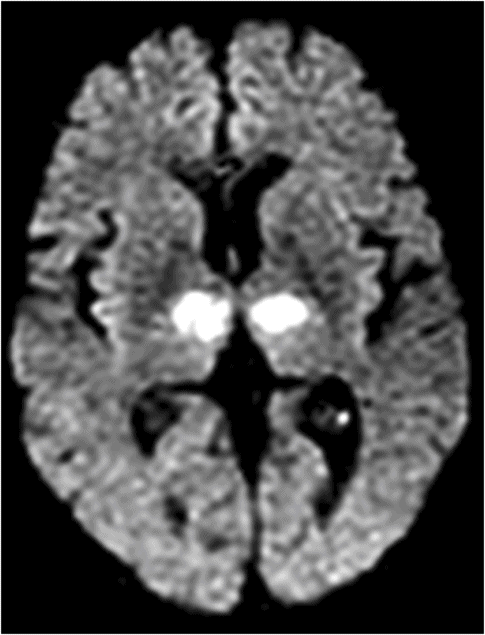
Diffusion-weighted magnetic resonance imaging shows bilateral paramedian infarction in a 67-year-old woman who presented with altered mentality, and rapid stupor. One day later, she became alert but showed persistent abulia, apathy, and severe amnesia as well as vertical gaze palsy.
Paramedian artery territory infarction may cause alterations in consciousness ranging from somnolence to coma [160]. Consciousness usually fluctuates and improves, but prolonged coma may result if the lesion extends into the midbrain tegmentum [157]. Approximately 50% of patients with bilateral lesions have persistent impairment of vigilance [160]. The initial stupor and subsequent hypersomnia are attributable to bilateral lesions in the intralaminar nuclei, which are part of rostral extension of the midbrain reticular activating system [131].
Anterograde amnesia frequently develops, but patients with paramedian thalamic infarction are known to present with less severe amnesia than those with tuberothalamic infarction [142]. The memory disturbance after paramedian thalamic infarction has been reported to be associated with damage to the intralaminar or dorsomedial nuclei of the thalamus. However, there is controversy as to whether damage to these nuclei can give rise to anterograde amnesia [152]. For instance, patients with a lesion affecting the intralaminar nuclei present with discrete amnesia but accompanied by severe distractibility, suggesting that the intralaminar nuclei are probably not memory structures per se [161]. Rather, coexisting damage to the anterior and dorsomedial nuclei may result in the most severe amnesia [162]. Therefore, memory disturbances associated with paramedian thalamic infarction could be explained by frontal dysfunction resulting from damage to dorsomedial and intralaminar nuclei [149]. Other behavioral abnormalities observed in paramedian thalamic infarctions are utilization behavior [163] and Kluver–Bucy syndrome, especially after bilateral paramedian artery infarction [164]. Rarely, a patient with right dorsomedial thalamic infarction presented as a paranoid schizophrenia-like psychosis [165], and impaired social cognition (i.e., perception of facial expressions of others and of sarcasm) was observed in a patient with bilateral dorsomedial thalamic infarction [166].
Inferolateral artery territory infarction
Ataxia and hypesthesia are the most common symptoms after inferolateral territory infarct [156], but behavioral changes and cognitive impairment can occur [152]. Patients with these lesions occasionally show executive dysfunction, including in planning, initiation, and regulation of goal-directed behavior [167], or aphasia [168].
Dementia associated with cerebral small vessel disease
Anatomy of cerebral small vessels
The penetrating small arteries of the brain are unique. The vessels forming the terminal branches from the major cerebral arteries divide and ramify in the pia mater to cortical and deep penetrating branches. The deep penetrating branches arise from the main artery penetrating into white matter to a depth of 3 or 4 cm perpendicularly. They are thin and long, lacking communications, thus constituting many independent small vascular systems. Unlike the main arterioles, the deep penetrating branches extend into the small lumen, thus making them sensitive to systemic hypertension [169].
Clinicopathologic correlates of small vessel disease and its radiologic manifestations
The small vessel pathologies manifest either lacunes or leukoaraiosis (white matter hyperintensieis in brain MRI) (Figures 19.15 and 19.16). Pathologically, lacunar infarction refers to a condition of small infarctions less than 15 mm in size resulting from occlusion of one single, deep-penetrating artery [169]. The lacunes are frequently located in deep gray nuclei (basal ganglia and thalamus), pons, and white matter of the centrum semiovale. White matter hyperintensities consist of periventricular white matter (anterior cap, rim or halo, and posterior cap) and deep white matter hyperintensities (Figure 19.16).
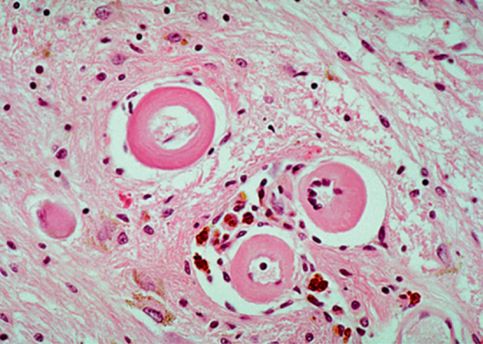
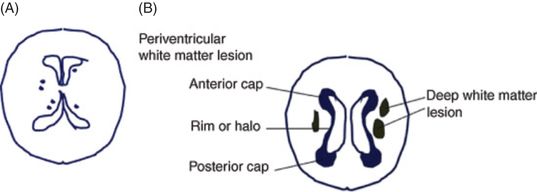
(A) Lacunar states in basal ganglia and thalamus. (B) Periventricular white matter and deep white matter.
The pathologic correlates of these white matter hyperintensities can be classified into [1] necrotic, black cystic lesions, isointense to CSF on T1 MRI and [2] non-necrotic, so-called “incidental” lesions, hyperintense to gray matter on T2 MRI. Necrotic lesions are often results of arteriopathies, such as intimal hyperplasia, atherosclerosis, arteriosclerosis, lipohyalinosis, amyloidosis, vasculitis, and CADASIL [170]. Supplied by deep penetrating arterioles from pial surface arteries with little collateral supply, periventricular white matter is particularly vulnerable to ischemia. Furthermore, arteriolar tortuosity, which appears around age 50, propelled by atherosclerosis and aging, requires higher perfusion pressure to supply white matter, increasing vascular resistance and resulting in chronic ischemia [171]. The mechanism of non-necrotic hyperintensities, which appear as focal, patchy, or confluent periventricular hyperintensities (PVH), is not clear. Arterial ischemia has been suggested as a culprit, but the focal loss of ependymal lining (“ependymitis granularis”) with axonal thinning, demyelination, and gliosis are other causative mechanisms [172].
Postmortem MRI and histopathologic studies suggest that a non-inflammatory, periventricular venulopathy with concentric collagen deposition correlates with PVH severity [173]. There is some evidence that this impaired interstitial fluid (IF) flow impedes the clearance of amyloid along perivascular spaces resulting in white matter hyperintensities in AD [174].
Overview of subcortical vascular dementia
Subcortical vascular dementia (SVaD), refers to the dementia resulting from ischemic lesions caused by small vessel disease [14–16]. There are two types of SVaD syndrome: one is dementia associated with predominant subcortical ischemic lacunes in deep nuclei (basal ganglia or thalamus) or the internal capsule, known as the lacunar type of SVaD, and the other is dementia associated with predominant ischemic changes in white matter, which is known as the white matter type of SVaD or Binswanger’s disease (subcortical arteriosclerotic encephalopathy) [175] (Figures 19.17 and 19.18).
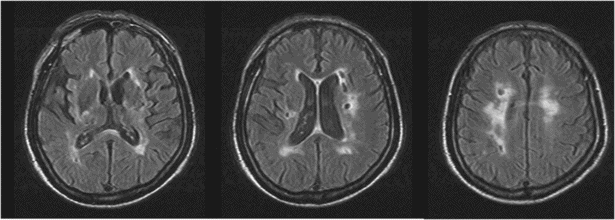
Subcortical vascular dementia with predominantly lacunar changes.
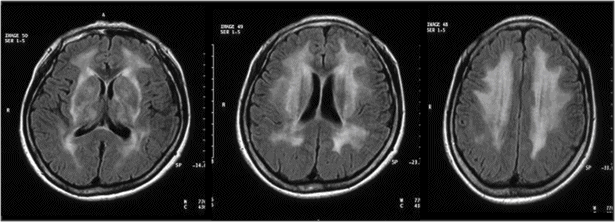
Subcortical vascular dementia with predominantly white matter changes, or Binswanger’s disease.
Lacunar state and Bingswanger’s disease commonly occur together because the underlying pathology involves the lenticulostriate and the penetrating subcortical arterioles of the hemispheric white matter simultaneously (Figure 19.19). This may explain why patients with lacunar strokes are more likely to have white matter changes [176] and to develop dementia [177] than those with other stroke subtypes.
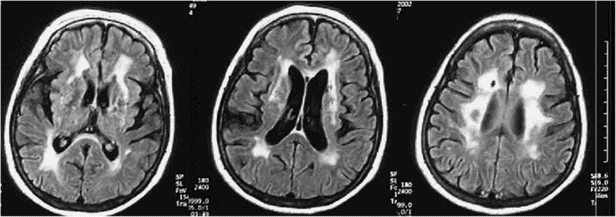
Subcortical vascular dementia with both mixed lacunar and white matter changes.
There has been suggestion that pure VaD may not be common and most patients with SVaD might have comorbidity of AD. However, recent studies suggest that pure SVaD is commoner than expected and has distinct anatomical substrates, and neuropsychological and neuropsychiatric profiles compared to Alzheimer’s disease dementia or mixed dementia. A PiB study in a group of severe WMH compatible with SVaD (AMPHIS study) showed 68.9% were negative for PiB and they were slightly younger, had better performance on the MMSE, and had a greater number of lacunes but less severe hippocampal atrophy on MRI than PiB positive SVaD [178], and more profound cortical thinning in bilateral inferior frontal, superior temporal gyri, and right medial frontal and orbitofrontal lobes compared to AD [179]. Another recent study has shown remarkable data on discriminating mixed dementia from pure SVaD by hippocampus and amygdala shape analysis with a discrimination accuracy of 82.4% (95.7% sensitivity and 75.6% specificity) when there is proven brain amyloid deposition in the PiB PET scan and with subcortical shape analysis using Free Surfer [180]. Cognitive changes of SVaD do overlap with AD; however, there are clear differences. Cognitive impairments in SVaD are related to ischemic interruption of frontal cortical circuits or disruption of cholinergic pathways that traverse the subcortical white matter [26].
The traditional cognitive screening test, the Mini Mental State Examination (MMSE), is biased toward detection of memory and language disturbance and, therefore, may not be sensitive in detecting the early presence of executive dysfunctions in SVaD [181]. Executive functions, planning and sequencing, speed of mental processing, performance on unstructured tasks, and attention tend to be disproportionately impaired in SVaD.
Memory impairment in AD is mediated by temporal areas; consequently, performance is poor in both recall and recognition. However, memory deficits in SVaD are more related to inattention through frontal executive dysfunction, leading to a retrieval defect pattern that is worse in recall but better on recognition and cued recall [182]. Recent PiB-PET studies confirmed the above findings that PiB negative SVaD had better memory but worse frontal function compared to AD [183]. Frontal involvement even in the early stage of SVaD also explains the fact that behavioral abnormalities in SVaD differ from those in AD. That is, patients with SVaD are more likely to show depression, agitation, and anxiety than those with AD. Therefore, it is important to recognize behavioral manifestations in SVaD for early detection of these diseases.
Neurological aspects of subcortical vascular dementia
Patients with lacunar states often have a history of abrupt onset and sometimes stepwise deterioration, as in MID. They tend to have more extensive medical histories of hypertension, and a greater likelihood of focal neurologic symptoms and signs compared with those with Binswanger’s type of SVaD [184]. Unlike most of the other vascular dementias, Binswanger’s disease often has an insidious onset with no significant lateralizing symptoms, and it may sometimes be mistaken for a degenerative disorder at the beginning of the disease process [185].
Once the disease has progressed, neurological manifestations of two groups do not differ. The neurological deficits associated with the two groups of SVaD can be divided into corticobulbar, corticospinal, and extrapyramidal dysfunctions. Corticobulbar dysfunctions include central facial palsy, dysarthria, and dysphagia. In addition to these symptoms, emotional ability or pathological laughing or crying with jaw jerk can occur, especially when the corticobulbar tracts are affected bilaterally. Corticospinal involvement manifests as motor weakness, asymmetrically increased deep tendon reflexes, and extensor plantar responses. When ischemic insult involves the extrapyramidal system, vascular parkinsonism occurs, which includes bradykinesia and rigidity, as well as small stepped gait (marche à petits pas), decreased arm swing, stooped posture, multistep turning, festination, and shuffling during walking [186]. Finally glabellar, snout, rooting, and grasp reflexes or frontal releasing signs are often present. A new motor scale, called the PEPS (Pyramidal and Extra Pyramidal Scale for sVCI), consisted of 34 items (for 60 total points) with five subdomains: corticospinal, corticobulbar, extrapyramidal signs, gait abnormalities, and gait severity. The PEPS had good inter-rater and test– retest reliability, and it correlated relatively with the UPDRS, NIHSS, MMSE, CDR, and ADL scales. An optimal cut-off score of PEPS to discriminate dementia or VaMCI from those without ischemia was 6.5 with a sensitivity of 88% and a specificity of 100%, showing that PEPS is a reliable and valid scale that can be used to assess and monitor motor impairment in patients with vascular cognitive impairment due to small vessel disease [187].
Neuropsychological aspects of subcortical vascular dementia
Frontal function, attention, and speed
The subcortical syndrome characterized by prominent dysexecutive syndrome, bradyphrenia, and mild memory deficits of retrieval defect pattern are the primary clinical manifestations of SVaD [182]. Of those frontal subcortical circuits, the anterior cingulate circuit and the dorsolateral prefrontal circuit play major roles in manifestation of SVaD symptoms [26, 188].
The impairment in executive function reflects the deficits in a number of cognitive functions, including attention or short-term memory (working memory), ability to plan a prospective action, and behavioral monitoring, and is responsible for a functional disability in everyday complex actions [189, 190, 191].
Patients with SVaD have significantly greater perseveration during tasks that assess set-shifting than during semantic testing [192]. A recurrent perseveration is believed to be the result of left temporal and parietal lobe pathologies and is associated with poor memory and language function, while a stuck-in-set perseveration is caused by frontosubcortical pathologies and is associated with the failure in mental set-shifting [193]. Unstructured tasks that require executive abilities, such as behavioral initiation, are also useful assessments. Other neuropsychological investigations suggest that deficits of frontal function can be identified before the onset of memory or other cognitive disturbances in patients with small vessel disease and ischemic injury to deep hemispheric gray and white matter structures. These findings suggest that changes in frontal functions occur well before the onset of memory problems in SVaD and this is a potential key to identifying patients at risk for developing VaD [194].
Memory and visuospatial function
Short-term memory, as estimated by digit span, has consistently been reported to be similarly affected in SVaD and AD [195]. The comparison of qualitative memory aspects in SVaD and AD has shown that both groups perform poorly on free recall of memory test. When free recall and recognition abilities are compared, patients with SVaD show better recognition memory than free recall, while those with AD show lesser efficacy on cued recall and impaired recognition than those with SVaD [195, 196]. This pattern is considered a retrieval defect pattern of memory, where patients are helped by semantic cues. Use of cued recall and recognition tasks significantly enhances the ability to discriminate SVaD from AD [197]. Better performances on recognition than recall also suggest that the underlying mechanisms of memory deficits in SVaD are problems of psychomotor slowing and retrieval more than storage problems. Visuospatial functions are reported to be better in SVaD than in AD, but overall specificity and positive predictive values are low [198].
Language function
Language has not been extensively studied in SVaD. Language tasks requiring semantics, including complex syntax comprehension and picture naming, are impaired in both AD and SVaD, but single-word repetition, oral reading of words and sentences, and fluency output is relatively spared in SVaD [199]. On confrontation naming, patients with SVaD are better on tests of naming, indicating preservation of semantic knowlege [196, 200], but those with SVaD manifest more perseveration in naming than those with AD [201].
Neuropsychiatric aspects of subcortical vascular dementia
There are more profound behavioral and affect changes in SVaD than in AD in most reports. Using the Neuropsychiatric Inventory (NPI) to explore behavior, those with cortical VaD and SVaD had higher mean composite NPI scores in all domains than those with AD [202, 203]. Their behavioral changes are characterized by depression, personality change, emotional bluntness, and psychomotor retardation. Subsequent studies also report that depression and anxiety are more common in SVaD than in AD [206]. Even when recognizing emotion, those with SVaD performed significantly worse than those with Alzheimer-type dementia on the emotion recognition task even when the cognitive status of each group did not differ [207].
Vascular mild cognitive impairment
The term vascular cognitive impairment (VCI) was first proposed as an umbrella term to emphasize the preventability of vascular-related cognitive dysfunction [2]. It comprises all types of vascular-related events. To minimize confusion related to the concept of MCI and for criteria utilized with mildly impaired groups, the term vascular MCI (VaMCI) is suggested by AHA/ASA for the group of patients with MCI of vascular origin [10].
The clinical importance of VaMCI is that as it could be the most prevalent form of cognitive disorder among those aged 65 to 84 years [208]; modifying the vascular risk factors and early drug treatment could prevent the progression of VaMCI to vascular dementia.
The new VaMCI criteria should include both a decline in cognitive function from a prior baseline and impairment in at least one cognitive domain within executive/attention, memory, language, and visuospatial function assessment and normal or mildly impaired instrumental activities of daily living as well. Probable VaMCI is reserved for VaMCI with evidence of cerebrovascular disease either with a clear temporal relationship between vascular event and onset of cognitive deficit or severity and pattern of cognitive impairment. Those VCI that do not meet the above criteria such as cognitive impairment due to mild subcortical small vessel disease, can be classified into possible VaMCI. Even though the AHA statement did not specify MR criteria for VaMCI of subcortical small vessel disease, it has been suggested by previous researchers. It was suggested for significant white matter changes in periventricular white matter (caps or rim) longer than 10 mm, and deep white matter consistent with extensive white matter lesion or diffusely confluent lesion ≥ 25 mm in T2 or FLAIR images [209, 210]).
Compared with patients with SVaD, those with svMCI were worse in all cognitive domains apart from recognition of Rey figures, alternating hand movement, and the Luria loop, where the two groups showed comparable performances [209]. In another study [210], when comparing neuropsychological performances between the svMCI group and the amnestic MCI (aMCI) group, the svMCI group performed less well in frontal executive function and the Rey copy task than the aMCI group, whereas the opposite was true for memory, which is consistent with the previous reports [211]. The burden of pure vascular disease such as lacunes was more associated with memory, frontal dysfucntions, and disease severity, but the total volume of white matter hyperintensities and the PibB retention ratio were associated only with memory dysfunction [212].
A neuropsychiatric study suggests that the presence of depression, albeit only in males, predicted conversion from subcortical vascular mild cognitive impairment to vascular dementia [213]. SvMCI with more lacunes in the frontal regions was associated with higher odds of depression, apathy, aberrant motor behavior, night-time abnormal behavior, and appetite changes, and larger WMH volumes in the frontal regions was associated with higher odds of apathy. Both were associated with a higher score of total NPI in contrast to an association of higher odds of delusions and irritability with greater PiB retention ratio [213]. When compared for cortical atrophy, svMCI showed cortical thinning in inferior frontal, orbitofrontal gyri, anterior cingulate, insula, superior temporal gyrus, and lingual gyrus compared to controls, while SVaD showed all these areas plus dorsolateral prefrontal and temporal cortices involvement [209]. These findings suggest a hierarchy exists between svMCI and SVaD and that svMCI defined according to the above criteria is a transitional stage between healthy controls and SVaD. Another study using 18F-FDG PET showed svMCI as a different disease from aMCI in terms of glucose metabolism [210].
Related posts:
 The non-fluent/agrammatic variant of primary progressive aphasia
The cognitive neurology of corticobasal degeneration and progressive supranuclear palsy
The non-fluent/agrammatic variant of primary progressive aphasia
The cognitive neurology of corticobasal degeneration and progressive supranuclear palsy
 Amnestic mild cognitive impairment
Amnestic mild cognitive impairment
 The Lewy body dementias
The Lewy body dementias
 Semantic dementia (semantic variant primary progressive aphasia)
Semantic dementia (semantic variant primary progressive aphasia)
 Autoimmune antibody-associated encephalopathy and dementia syndromes
Autoimmune antibody-associated encephalopathy and dementia syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


