Fig. 5.1
Schematic representation of the renin–angiotensin pathway. Angiotensinogen (AGT) cleaved by renin to produce angiotensin I, which is converted to angiotensin II through ACE. Ang II binds to AT1R and AT2R, inducing vasoconstriction and vasodilation, respectively. Both Ang I and II can be then degraded by peptidases, leading to the production of several Ang metabolites including Ang III, Ang IV, Ang-(1–7), Ang A, Angioprotectin and Alamandine, which exert various physiological effects through their respective receptors. Drugs that antagonize the RAS include ACEi, which reduce the formation of Ang II, and ARBs, which antagonize the actions of Ang II meditated primarily by AT1R. ACE angiotensin converting enzyme, ACEi angiotensin converting enzyme inhibitor, AGT angiotensinogen, Ang angiotensin, APA aminopeptidase A, APN aminopeptidase N, ARB angiotensin receptor blocker, ATR angiotensin receptor, MASR Mas receptor, MrgDR Mas-related G-protein coupled receptor
1.2 Hypertension Induced by Ang II
1.2.1 The Renin–Angiotensin System (RAS)
The RAS/Ang II cascade is a complex enzymatic pathway that involves the conversion of an inactive pro-hormone, angiotensin I (Ang I), into several active peptides involved in the regulation of BP, homeostasis, hormone secretion, and behavioral and cognitive function [33, 34] (Fig. 5.1). The RAS is initiated by renin, an enzyme secreted subsequently to systemic hypotension or certain nerve impulses, and which converts the precursor protein angiotensinogen into Ang I. The latter is, in turn, converted by ACE into the octapeptide Ang II, which is considered the primary effector hormone of the RAS. Both Ang I and Ang II can be then degraded by peptidases, leading to the production of several Ang metabolites including angiotensin III (Ang III), angiotensin IV (Ang IV), angiotensin 1–7 (Ang-1–7), angiotensin A (Ang A), angioprotectin, and alamandine [35] (Fig. 5.1). This cascade is present in brain and in the endothelium of most blood vessels where it is known for its hypertensive effect and its ability to stimulate vascular remodeling. As such, Ang II plays an important role in the pathogenesis of HT and in cognitive function. The action of the RAS/Ang II cascade on behavior is likely, at least in part, mediated by HT-induced cerebrovascular changes [36]. Recent evidence suggests that the brain RAS is involved in cerebrovascular/cardiovascular regulation [37], neuronal plasticity [38], cognitive function, and dementia [39].
Pharmacologically, all the angiotensin-derived peptides are active and induce various physiological effects, suggesting the presence of multiple angiotensin receptors. Indeed, several Ang receptors have been identified, such as the Ang receptor types 1–4 (AT1R–AT4R), Mas, and MrgD receptors [40].
The most common Ang II receptor is the AT1R, which is expressed in various tissues such as in the gut, heart, blood vessels, kidney, and brain [41]. The actions of Ang II mediated via AT1R include vasoconstriction, endothelial dysfunction, oxidative stress, smooth muscle hypertrophy, and chronic inflammation [42]. Notably, AT1R can also be activated by some Ang metabolites such as Ang A or Ang III (Fig. 5.1) however with lower affinity [43].
The Ang II/AT1R effects can be counterbalanced by activation of AT2R or AT4R. AT2R has 34 % homology with AT1R, is expressed ubiquitously in the fetus, and decreases rapidly after birth though it can be strongly upregulated following tissue injury. Activation of AT2R in animal models of ischemia is protective, as it reduces the infarct area by increasing cerebral perfusion, decreases superoxide production, and promotes neuronal cell differentiation and neurite growth, thereby leading to neuronal repair, and reduced axonal degeneration and inflammation. Further, AT2R seems to exert a hypotensive effect, and is thought to reduce the affinity of AT1R for Ang II [42].
In addition to its direct effects on AT2R, Ang II can counter the vasoconstrictor effects of AT1R through its bioactive metabolites. For instance, Ang III, a breakdown product of Ang II by aminopeptidase A, has been found to mediate coronary vasodilatation [44] via AT2R activation. Other Ang II metabolites may exert similar vasodilatatory effects, such as Ang-(1–7) and angioprotectin, both of which bind to the Mas receptors [43], or Ang IV and alamandine through their respective actions on AT4R [45] and MrgD receptors [35].
1.2.2 Short- and Long-Term Administration of Ang II
The role of Ang II has been widely studied and implicated in learning and memory in various animal models using a diversity of tests. These include associative learning measured by passive and conditioned avoidance tasks as well as mazes and object recognition tests commonly used to evaluate spatial learning and recognition memory. Here, we will describe the effects of acute and chronic Ang II treatment on animal models.
In an acute model of intracerebroventricular (ICV) injection, rats administered with Ang II at doses of 0.1, 0.5, and 1 μg 15 min before training sessions showed facilitated learning and retention in the conditioned avoidance test [46]. These beneficial effects of Ang II were blocked by administration of the nonselective ARB saralasin [46], suggesting that Ang II through activation of brain angiotensin receptors improves learning and memory. The work of Braszko and colleagues [47] further showed that ICV administration of the ARB losartan (1 nmol, 15 min prior to training) abolished the ability of Ang II-(3–7) to enhance object recognition in rats even though no beneficial effect of Ang II-(3–7) was observed in the conditioned avoidance test. However, it is uncertain whether these effects were mediated by AT1R, as Ang II-(3–7) has a considerably lower affinity at this receptor compared to Ang II [47].
In contrast, other studies suggest that Ang II injection into the neostriatum or dentate gyrus (DG) of the hippocampus decreases memory and learning capacity [48, 49]. Indeed, the injection of 5 ng of Ang II into the DG administered 1.5 h prior to training impairs retention in the conditioned avoidance task. These effects were reversed by the ARB losartan in a dose-dependent manner, suggesting a role for AT1 receptors (see Sect. 3) [48, 49]. The aforementioned studies have used acute ICV injections of Ang II, which have been associated with several drawbacks that could impede the interpretation of results. Indeed, such injections increased BP and heart rate that are anxiogenic and associated with a wet dog shake behavior [50]. In contrast, injections in the DG are restricted to this area and act within the BBB.
Interestingly, a recent study showed that chronic administration (3 weeks) of Ang II with an osmotic pump impairs cognitive function [51] whereas a single ICV injection 15 min before training improves memory [46], suggesting that Ang II can have different actions on behavior depending on the period of exposition. All these findings highlight the complex and multifactorial actions of RAS in learning and memory.
1.3 Transverse Aortic Constriction
The Transverse aortic constriction (TAC) model is primarily used to create chronic heart failure, but it has also been associated with HT from the right carotid artery to the brain [52]. TAC consists of constriction of the aortic lumen between the innominate artery and the left carotid artery, which results in pressure overload-induced cardiac hypertrophy and heart failure [53] (Fig. 5.2). Rockman and colleagues were the first group to validate the murine TAC model, and since 1991 it has been widely used as a reliable tool to mimic human cardiovascular diseases in order to investigate the mechanisms involved in heart failure. The TAC model creates a pressure gradient across the aortic arch and the pathology evolves with age. Work from the Lembo group showed that TAC mice display decreased blood flow contralateral to the TAC (left carotid artery) associated with low peak velocity of cerebral blood flow between (CBF) 1 and 28 days [54, 55]. As a consequence, the peak velocity in the left carotid artery is decreased and returns to baseline levels 14 days after TAC surgery [54]. Interestingly, the day after the surgery, blood flow in the right carotid artery is twice that of the left artery [53] (Fig. 5.2). Moreover, brain injury in TAC mice was evidenced in hippocampus and cortex, two areas related to cognitive function. In this respect, it is quite surprising that only one group has investigated the effects of TAC on cognition [56].
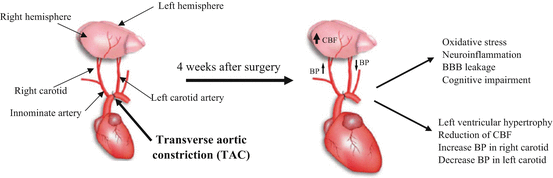
Fig. 5.2
Transverse aortic constriction (TAC) mouse model. Transverse aortic constriction (TAC) consists of constriction of the aortic lumen between the innominate artery and the left carotid artery. After 4 weeks, TAC induces hypertension with hypertrophy of the left ventricle, enhancement of CBF in right hemisphere but reduction in the left hemisphere, and increased blood pressure (BP) in right carotid with concurrent BP decreases in the left carotid artery. Moreover, TAC induces oxidative stress, neuroinflammation, BBB leakage, and cognitive impairment. Modified with permission from [172]
Indeed, Carnevale and colleagues reported that TAC mice display cognitive deficits in the novel object recognition test and in spatial memory measured in the Morris water maze [56]. Furthermore, they found that blockade of receptor for advanced glycation end products (RAGE) in the brain vasculature had a preventive effect on cognitive deterioration, and that RAGE knockout mice with TAC surgery were protected from cognitive failure. This study was the first to demonstrate that activation of cerebrovascular RAGE, which transports amyloid-β (Aβ) peptide from blood to brain, can induce cognitive dysfunction possibly through increased oxidative stress [57]. Indeed, another group demonstrated that TAC triggers oxidative stress and inflammatory responses that can favor neurodegeneration [53], and these effects have been related to BBB dysfunction [53]. In this respect, the work of Carnevale and colleagues highlights the importance of chronic vascular insults in triggering cognitive dysfunction by altering cerebral blood flow (CBF) (Fig. 5.2) and increasing oxidative stress, neuroinflammation, and BBB leakage [56]. Hence, this model can be a good tool to evaluate whether chronic HT can exacerbate cerebrovascular dysfunction and precipitate cognitive decline in individuals with increased risk factors for dementia (see Table 5.1).
Table 5.1
The effect of RAS-acting drugs on AD pathology and cognitive function: recent findings from AD experimental models
Study | ACEi tested (dose-duration) | In vivo model of AD (age at first dose) | Effect on AD pathology and cognitive function |
|---|---|---|---|
1. | Captopril (20 or 25 mg/kg/day—6 months) | Tg2576 (12 months) | • Normalized AD-related increase in ACE levels and activity |
• ⇓ Hippocampal Ang II accumulation and ROS generation | |||
• ⇓ Amyloidogenic processing of APP by β and γ secretases | |||
• ⇓ Aβ accumulation in brain | |||
2. | Captopril (30 mg/kg/day—7/11 months) | Tg2576 (6 months) | • ⇑ Aβ42 (but not Aβ40) |
3. | Enalapril (10 mg/kg/day—1 week) | Injection of Aβ1-40 for 2 days in ICR mice (7 weeks) | • No effect on Aβ1-40-induced activation of hippocampal microglia and astrocytes |
Imidapril (3 mg/kg/day—1 week) | • No effect on cognitive decline | ||
Perindopril (1 or 3 mg/kg/day—1 week) | Injection of Aβ1-40 for 2 days in ICR mice (7 weeks) | • Prevented Aβ1-40-induced activation of hippocampal microglia and astrocytes | |
• ⇓ Hippocampal oxidative stress caused by iNOS induction and extracellular superoxide dismutase downregulation | |||
• Prevented cognitive decline | |||
Perindopril (1 mg/kg/day—4 weeks) | PS2 APP Tg (3 months) | • Suppressed hippocampal astrocyte activation and ⇓ superoxide | |
• No effect on brain Aβ deposition | |||
• Prevented cognitive decline | |||
4. | Enalapril (1, 3, or 10 mg/kg/day—5 days) | Injection of Aβ25-35 in ICR mice (5–6 weeks) | • Only Perindopril reversedcognitive decline, perhaps as it was the only one able to inhibit brain ACE activity by >50 % |
Imidapril (0.3, 1, or 3 mg/kg/day—5 days) | |||
Perindopril (0.1, 0.3, or 1 mg/kg/day—5 days) | |||
5. | Lisinopril (10 or 15 mg/kg/day—14 days) | Injection of streptozotocin (STZ) in swiss albino mice (20–30 g) | • Both lisinopril and telmisartan ⇓ STZ-induced learning and memory impairment, and biochemical changes |
Telmisartan (5/10 mg/kg/day—14 days) | • Effects were abolished with PPAR-γ antagonist (BADGE) | ||
6. | Telmisartan (pretreatment–0.35 mg/kg/day—2 weeks) | Injection of Aβ1-40 for 2 weeks in ddY mice (8 weeks) | • ⇓ Aβ deposition and ⇑ CBF |
• Prevented cognitive decline | |||
• Effects were abolished with PPAR-γ antagonist (GW9662) | |||
7. | Telmisartan (pretreatment—100 mg/kg/day—2 weeks) | Injection of Abeta1-40 for 2 weeks in ddY mice (8 weeks) | • ⇓ Aβ deposition by 48 % |
• Prevented cognitive decline | |||
• Effects were abolished with PPAR-γ antagonist (GW9662) | |||
Losartan (pretreatment—100 mg/kg/day—2 weeks) | Injection of Abeta1-40 for 2 weeks in ddY mice (8 weeks) | • No effect on Aβ deposition | |
• Prevented cognitive decline | |||
• Not affected by PPAR-γ antagonist (GW9662) | |||
8. | Losartan (10 mg/kg/day—3 months) | APP J20 mice (15 months) | • Normalized cerebral glucose uptake induced by whisker stimulation (neurometabolic coupling), neurovascular coupling, and cerebrovascular reactivity |
• Rescued cognitive deficits | |||
• ⇓ Astrogliosis and oxidative stress | |||
• No effect on soluble Aβ species and Aβ deposition in brain | |||
• No effect on cortical cholinergic innervation | |||
Losartan (~10 mg/kg/day—10 months) | APP J20 mice (2 months) | • Normalized cerebrovascular reactivity | |
• Prevented cognitive decline | |||
• No effect on brain soluble Aβ species and Aβ deposition | |||
9. | Valsartan (10 mg/kg/day—5 months) | Tg2576 mice (6 months) | • ⇓ Cognitive decline |
• ⇓ Aβ oligomerization into high-molecular weight oligomeric peptides, involved in cognitive deterioration | |||
• No effect on CM-IDE activity and total Aβ1-42 | |||
Valsartan (40 mg/kg/day—5 months) | Tg2576 mice (6 months) | • ⇑ Cortical activity of CM-IDE, involved in Aβ clearance | |
• ⇓ Aβ1-40 and Aβ1-42 |
2 Part II: Cerebrovascular Pathology and Dementia
HT is a risk factor for various disorders such as stroke, atherosclerosis, myocardial infarction, and vascular diseases. Although HT is known to induce vascular dementia (VaD) [58], recent reports have further highlighted its role in the development of Alzheimer’s disease (AD) [59]. Here, we will focus on the effects of HT on cerebrovascular pathology and how it can be linked to dementia. First, common pathogenic mechanisms to HT, VaD, or AD will be described and then, in the last part of the review, the effects of antihypertensive therapies in animal models of cognitive impairment will be discussed.
2.1 Definition of Vascular Dementia (VaD) and Alzheimer’s Disease (AD)
High BP is a major risk factor for cognitive impairment and, ultimately, for dementia. The definition of dementia on vascular bases has been suggested by Hachinski and colleagues [60], and inspired from the work of Tomlinson [61]. VaD may result from ischemic or hemorrhagic infarcts, or from cardiovascular or circulatory disturbances that affect cerebral regions important for memory, cognition, and behavior [60]. Vascular cognitive impairment (VCI), on the other hand, is a recent notion that encompasses the entire spectrum of cognitive dysfunctions associated with all types of cerebrovascular injury, with VaD as its most severe form. However, this category is typically used to designate patients with risk factors for cardiovascular diseases and with cognitive impairment insufficient to induce a loss of autonomy [62, 63]. VaD may coexist with many cerebral and systemic disorders that can affect cognitive function in older populations, especially AD. In fact, the coexistence of VaD and AD is termed mixed dementia, a diagnosis, which can be based on clinical evidence or the presence of ischemic lesions. The boundaries between VaD and AD are not clear, and it is not always possible to determine whether the cognitive loss is strictly caused by AD or mediated by vascular factors [64–66]. Effectively, while AD has been widely recognized as the predominant form of dementia in the elderly since the 1980s, the majority of old autopsied subjects with dementia present the typical lesions of AD in the hippocampus in addition to cerebrovascular lesions. Furthermore, 20 % of cases of pathologically confirmed AD were not clinically demented. It has been suggested that vascular lesions are needed for the clinical expression of AD [67]. Accordingly, the nun study [68] found that at early stages of AD, lacunes or lacunar infarcts increase more than 20 times the risk of clinical expression of dementia. The literature thus increasingly suggests that cerebrovascular lesions constitute the most important cause of dementia in the elderly. Therefore, it is important to define VaD based on vascular factors that lead to cognitive dysfunction.
It is well accepted that VaD is associated with vascular diseases and is characterized by cerebral hemorrhages, ischemic infarcts, lacunes, white matter (WM) lesions, BBB dysfunction, and/or microvascular degeneration [65]. These cerebrovascular alterations are thought to contribute to the development of cognitive impairment by altering neuronal networks involved in memory, cognition, behavior, and executive functions [69]. In contrast to VaD, AD is a complex and multifactorial neurodegenerative disorder resulting from various factors. In addition to the cerebral amyloid angiopathy (CAA), HT is also thought to contribute to the onset and evolution of cerebrovascular and cognitive impairments in AD, likely through its deleterious effects on the brain vasculature (for a review, see [70]). Indeed, HT can affect cerebrovascular function at multiple levels, which will then alter normal brain function.
2.2 The Neurovascular Unit in HT, a Role in Cognitive Failure
HT is a major modifiable risk factor for cognitive decline in the elderly that can lead to VaD or AD, or a combination of these disorders [71–73]. The neurovascular unit (NVU), which is the association between (a) vascular cells (brain endothelial cells, pericytes, and vascular smooth muscles cells (VSMC)); (b) glial cells (astrocytes); and (c) neurons, plays key roles in the functioning of a healthy brain. Components of the NVU interact closely to regulate the CBF in response to regional changes in neuronal activity, a phenomenon termed neurovascular coupling (NVC). Moreover, the NVU is also involved in BBB permeability, cell matrix interactions, neurotransmitters turnover, toxin removal from the brain, as well as angiogenesis [74]. Hence, alterations in any of the cellular compartments of the NVU will impinge on its good functioning. In VaD and AD, the NVU is altered, which results in impaired increases in activity-evoked CBF during cognitive tasks [69, 70, 74–76].
For example, it has been observed that anatomically, SHRs have morphological differences such as decreased brain weight and volume compared to their normotensive counterparts [77–79]. Comparable neuronal density in SHR and WKY rats at 6–7 months of age has been reported in one study [78]. However, in specific brain areas such as the periventricular preoptic nucleus, paraventricular nucleus, arcuate nucleus, ventromedial and anterior hypothalamus, organ vasculosum lamina terminalis, and subfornical organ, it has been observed that the size and number of neurons are reduced in SHRs compared with their normotensive cohorts [79, 80]. The hippocampus, which is the key region for learning and memory, is smaller in 6-month-old SHRs compared to age-matched normotensive WKY rats [4, 6, 77, 81]. Reduced neuronal density in the CA1 region of the hippocampus has also been documented in SHRs at 14 weeks of age [79]. As observed in humans with AD or VaD using neuroimaging techniques, WM alterations occur often and their incidence increases with aging and HT [82, 83]. WM lesions are often associated with cognitive deficit and gait disorders in the elderly and could explain the correlation of cerebrovascular disease and the development of cognitive impairment [84].
In fact, HT alters cerebrovascular morphology causing inward hypertrophy, a type of vascular remodeling. The main feature of the inward hypertrophy is thickening of the arterial wall and reduction in lumen diameter [85–88]. In addition, high BP alters CBF regulation (see Chap. 6). Consequently, HT might lead to a marked reduction in WM blood flow.
Cognitive deficits in HT may result from alterations of the cerebral circulation [89, 90]. Incidentally, Ang II plays an important role in the cerebral vasculature and the pathogenesis of HT, as discussed in previous sections (1.2.1 and 1.2.2). HT, mainly through Ang II, increases oxidative stress that alters vessel structure and function [91], which in turn can affect CBF regulation. Ang II produces cerebrovascular remodeling, promotes vascular inflammation, and impairs CBF regulation [92–96]. In addition, both short- and long-term administrations of Ang II impair NVC [37, 97–103]. The vasodilator ability of the vessels is thus impaired, and therefore brain perfusion is diminished and probably influences the cognitive function of hypertensive patients. Antihypertensive drugs, including angiotensin converting enzyme inhibitors (ACEis) [104, 105] and ARBs [106–109], improve cerebral perfusion in hypertensive patients. Recent investigations suggest that ACEis are not as powerful as ARBs in preventing the decline of CBF seen in elderly hypertensive subjects [109], suggesting that ARBs may provide additional benefits. This could be due to the consequent decrease in kinin synthesis or the production of Ang II metabolites that exerts protective effects on the brain (Fig. 5.1).
2.3 Dysfunction of BBB in HT, Relevance to VaD and AD
The BBB prevents the free diffusion of circulating molecules, leukocytes and red blood cells into the brain interstitial space, maintains a homeostatic environment for neurons and glial cells, and helps clearing metabolites from the brain. The BBB is formed by the presence of high resistance tight junctions that fuse brain capillary endothelial cells together into a continuous layer, these cells being endowed with highly selective transporter systems [76]. BBB breakdown has been associated with cerebrovascular disease and HT [76].
There is growing evidence that HT has deleterious effects on BBB permeability. For example, it has been shown that BBB permeability in Ang II-dependent hypertensive mice (Ang II-infused wild-type mice) is increased compared to vehicle-treated mice [110, 111]. There is emerging evidence that Ang II-dependent HT affects BBB function even though the mechanism remains unclear. Indeed, some BBB transporters have been highlighted for their possible contribution in the pathogenesis of AD. Particularly, RAGE and the low-density lipoprotein receptor-related protein-1 (LRP1) have been involved in the altered transit of Aβ peptide in and out of the brain in AD [76]. A faulty clearance of Aβ due to impairments in these BBB transporters has been associated with Aβ accumulation in brain parenchyma and blood vessels [112]. It has also been shown that the expression of RAGE is augmented in both neurons and endothelium of AD patients or mouse models, which would further increase Aβ-induced pathogenic responses [113–115]. Moreover, as mentioned above, the group of Lembo showed that blockade of RAGE in the brain vasculature protects the cognitive deficits in TAC mice [56]. In opposition, LRP-1, a member of the LDL receptor family that regulates transport of tissue plasminogen activator, apolipoprotein E, amyloid precursor protein (APP), and Aβ is downregulated in normal aging and in AD brains [116]. Because the LRP knockout model is lethal [117], receptor-associated, protein-deficient knockout mice (RAP−/−) known to present reduced LRP levels [118, 119] were crossed with mice overexpressing mutant human APP. This model exhibits significantly enhanced cerebral Aβ pathology with increased cerebrovascular and parenchymal Aβ plaques compared to control mice [120]. Similarly, the TAC model of HT presents an “AD-like pathology,” in which the features of VaD such as neuroinflammation, Aβ deposition, and cognitive deficits are exacerbated [56].
2.4 Dysregulation of CBF in HT, Relationships with VaD and AD
In hypertensive patients, it has been reported that dysregulation of CBF results possibly from several factors such as aging, vascular alterations, and HT [121–123]. Indeed, using resting-state PET, it has been found that CBF is reduced in the prefrontal, anterior cingulate, and occipital cortices in humans [124]. These areas are important for memory [125, 126] and executive function [127]; attention and error monitoring [128]; and visual perception and object recognition [129, 130], respectively. This suggests a pattern of functional vulnerability to HT that may account for the accelerated cognitive decline in hypertensive individuals compared to normotensive controls [124]. In accordance with these findings, Ang II administration in mice for 30 min or 7 days impairs the increase in CBF induced by whisker stimulation, which activates the somatosensory cortex [97], an effect not attributed to high BP. The effect of HT on CBF changes will be described in more detail in Chap. 6, but we can highlight that HT might exacerbate the cerebrovascular dysfunctions observed in animal models of AD.
2.5 Impairment of Adult Neurogenesis in HT, Relevance to Cognitive Dysfunction
Adult hippocampal neurogenesis refers to the process whereby new granule cells are generated from a population of neuronal progenitor cells residing in the subgranular zone of the DG [131, 132]. These newborn neurons migrate into the granule cell layer of the DG where they differentiate into granular cells [133, 134] and integrate into pathways intimately associated with spatial learning and memory [135], which suggests that DG neurogenesis might influence these functions. Recently, new granular cells have been shown to exhibit high levels of long-term potentiation (LTP) and to modulate memory encoding [136], stability, and clearance during infancy and adulthood [137, 138]. Several lines of research suggest an association between HT-induced altered hippocampal neurogenesis and cognitive impairment. Indeed, longitudinal studies revealed high BP in mid-life to be correlated with hippocampal atrophy and cognitive impairment later in life [131, 132, 136, 137]. Furthermore, studies have shown that hippocampus-associated behaviors, such as learning, memory, attention, and locomotor activity, are impaired in both SHRs [139] and in mice with Ang II-induced HT [51]. SHRs also display a range of alterations in the hippocampus ranging from reduced volume [4, 6, 77, 81] and neuronal density in the CA2 region [78] to changes in the morphology of pyramidal neurons [140]. Surprisingly, neurogenesis was found to be enhanced in SHRs as indicated by increased levels of doublecortin immunoreactivity [141], a marker of immature neurons, supporting the idea that hypertensive brain damage is accompanied by repair processes.
3 Part III: Potential Benefits of Antihypertensive Drugs on Cognitive Impairment
It is well known that VaD and AD share similarities and can overlap in many patients, and HT is a major risk factor for both pathologies [58, 59]. As mentioned previously (Sect. 2.1), a clear diagnosis of VaD, AD, or mixed dementia is sometimes difficult, and antihypertensive drugs have shown benefits in reducing the incidence of developing both VaD and AD [59, 142, 143]. For example, hypertensive patients treated with ACEis that block the conversion of Ang I to Ang II or with ARBs that primarily antagonize the action of Ang II at AT1R (Fig. 5.1) are less susceptible to develop dementia of either type [144–148]. The mechanisms responsible for these protective effects of antihypertensive drugs have not been identified, but recent progress in animal models has yielded new avenues of investigation (Fig. 5.3).
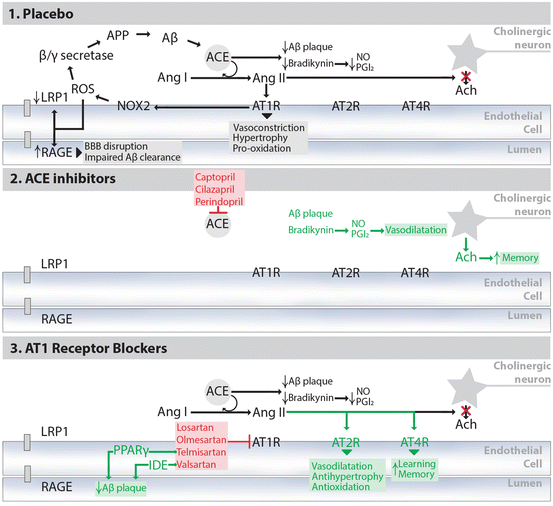
Fig. 5.3
Hypothetical mechanisms underlying the effects of RAS-acting drugs on pathology and cognition in experimental models of AD. Conflicting evidence with regard to the effect of ACEis and ARBs on AD pathology and cognition suggests a complex molecular interplay between the RAS and the pathogenesis of this disease, which is illustrated here in three different experimental conditions: administration of placebo, ACEis, or ARBs. (1) In AD, ACE exerts protective effects by inhibiting Aβ aggregation and toxicity [149]. However, these beneficial effects may be overridden by the establishment of a vicious cycle of ACE-dependent generation of Aβ [156]. Moreover, by generating Ang II, ACE causes Ang II/AT1R-mediated ROS generation, which may disrupt the BBB and impair Aβ clearance [112]. It also leads to the inhibition of ACh release [173], and to various physiological effects associated with HT, such as vasoconstriction, hypertrophy, and pro-oxidation [42]. (2) Brain-penetrating ACEis (e.g., captopril, cilazapril, and perindopril), by inhibiting ACE, block the beneficial effects of ACE-mediated Aβ degradation. However, they also increase ACh levels, possibly leading to improved memory; they block the pathological effects downstream of Ang II/AT1R activation, such as ROS generation and BBB disruption; and they block the degradation of bradykinin, a potent vasodilator which mediates its effects through NO and PGI2. (3) With ARBs, the beneficial Aβ-degrading function of ACE is preserved, and the specific blockade of AT1Rs results in a more complete inhibition of Ang II/AT1R-mediated effects. As such, circulating Ang II is free to bind to AT2R, which is associated with many protective effects, or to be metabolized to Ang IV, which may improve learning and memory through the binding and inactivation of AT4R [169]. Furthermore, the ARB telmisartan exerts additional positive effects on AD pathology through PPARγ activation [163, 174, 175], and the ARB valsartan when administered at high doses increases the activity of cortical IDE, which is involved in Aβ clearance [164]. Based on these hypothetical pathways, ARBs may confer additional protective effects for AD compared to ACEis although this remains to be investigated in future studies
3.1 The Effects of Angiotensin Converting Enzyme Inhibitors
Much controversy has surrounded the role of ACE in AD pathogenesis. Indeed, while many in vitro studies have shown ACE to promote degradation and clearance of Aβ [149, 150], possibly by cleavage at its Arg5-His6 site [151], ACEis have produced positive effects on cognition in various AD models [152, 153] (Table 5.1), suggesting that Ang II may play a role in the pathogenesis of AD. Effectively, ACE can be induced by aggregated Aβ both in vitro and in vivo [154, 155], and it is possible that the resulting increased production of Ang II in AD models could mediate pathogenic effects that would override the benefits of ACE-mediated Aβ clearance (Fig. 5.3 (part 2)). Indeed, positive effects of ACEis on AD pathology have been demonstrated. Abdalla and his group administered captopril in the drinking water of Tg2576 APP mice starting at 12 months of age and found delayed signs of neurodegeneration by six months [156]. This was accompanied by reduced amyloidogenic processing of APP, and decreased ACE activity and reactive oxygen species (ROS) production in the hippocampus [156] (Fig. 5.3 (part 2)). However, a previous study found the long-term use (7 or 11 months) of captopril administered in the diet initiated at 6 months of age in Tg2576 APP mice to be associated with an increased Aβ42 deposition [157]. These apparent discrepancies may be attributed to differences in the methodology, treatment duration, or age of the animals when initiating the treatment, emphasizing the need for further studies. Hirawa and colleagues observed that long-term treatment (18 months) with the ACEi cilazapril improved memory in aged hypertensive DS rats in the passive avoidance task, concurrently with increases in neuronal cell and capillary densities in the CA1 region of the hippocampus, a key area for memory [32].
Related posts:
 Hypertension and Brain Inflammation: Role of RAS-Induced Glial Activation
Hypertension and Brain Inflammation: Role of RAS-Induced Glial Activation
 A Systematic Review of Reviews to Examine the Association Between Hypertension and Cognitive Impairments in Adults
A Systematic Review of Reviews to Examine the Association Between Hypertension and Cognitive Impairments in Adults
 Arterial Stiffness and the Brain
Arterial Stiffness and the Brain
 Endothelium, the Blood–Brain Barrier, and Hypertension
Endothelium, the Blood–Brain Barrier, and Hypertension
 Hypertension and Stroke
Hypertension and Stroke
 Neuroimaging as a Research Tool in Human Essential Hypertension
Neuroimaging as a Research Tool in Human Essential Hypertension
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


