Symptom
Dementia
MND
ExPy
Accumulated protein
α-Synuclein
DLB
PD, MSA
Aβ
AD
Tau
AD
FTD
PSP, CBD
TDP-43
FTD
ALS
FUS
FTD
ALS
The components of ubiquitin-positive inclusions in ALS and FTD were not identified until 2006. In 2006 TDP-43 was finally identified as a component of ubiquitin-positive inclusions in ALS and FTD (Neumann et al. 2006; Arai et al. 2006), suggesting the concept of TDP-43 proteinopathy (Neumann et al. 2007). The system and cell type involved in each disorder are correlated with the unique clinical phenotype (Fig. 1.1). For example, the behavior variant of FTD (bvFTD), which is characterized by prominent early personality or behavioral changes associated with frontotemporal lobar degeneration (Rascovsky et al. 2011), shows inclusions comprising several different proteins. Of 66 cases of bvFTD, 42 % had tau-immunopositive inclusions, 30 % had TDP-43-immunopositive inclusions, and 13 % had FUS-immunopositive inclusions (Chare et al. 2014).
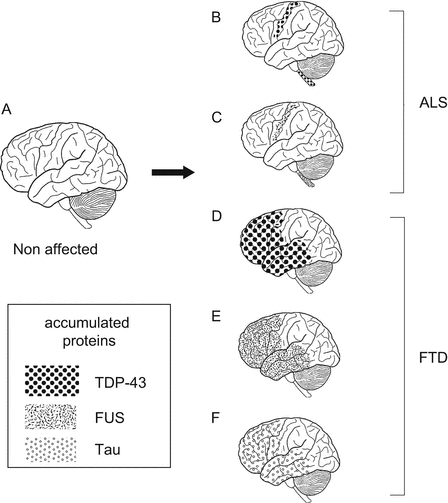
Fig. 1.1
Abnormal protein accumulation in conformational disease: (A) non-affected brain; (B, C) ALS-affected brains with (B) TDP-43 or (C) FUS protein inclusions in the pyramidal tract, motor cortex, and spinal cord; (C–E) FTD-affected brains with (D) TDP-43, (E) FUS, or (F) tau protein; (B–E) abnormal protein can be deposited in multiple lesions and can bring about several different diseases; (D–F) several proteins can accumulate in one particular system independently and can produce a clinically indistinguishable disease
1.3 The Initiation of Conformational Disease
Evidence for inclusion bodies defining specific neurodegenerative disorders has led to the concept of conformational disease caused by proteins unfolding (Carrell and Lomas 1997) such that they lose their normal three-dimensional structure and develop a tendency to aggregate. Basically, the proposed process responsible for aggregate formation is one in which the conformation of a monomeric protein is altered through abnormal phosphorylation, truncation, and/or change to a β-sheet structure. These proteins then form dimers, followed by oligomers and fibrils, which are insoluble (Barghorn et al. 2004).
Several mechanisms have been proposed for initiation of the disease process. First, in hereditary neurodegenerative disease, the mutant form of the disease-related protein acquires aggregability. Polyglutamine diseases have been well studied as models for conformational disease (Orr and Zoghbi 2007). Polyglutamine diseases are hereditary neurodegenerative diseases that include HD, spinocerebellar ataxia (SCA) 1, 2, 3, 6, 7, and 17, dentatorubral-pallidoluysian atrophy (DRPLA), and spinal and bulbar muscular atrophy (SBMA). They are caused by expansion of a polyglutamine stretch in the causative gene (Orr and Zoghbi 2007; Takahashi et al. 2010). In these disorders, inclusions that are immunopositive for the expanded polyglutamine stretch are observed in the affected neurons (Nagai et al. 2007). Although there is no functional relevance in these causative proteins, the length of the polyglutamine stretch is closely correlated with age at disease onset and disease severity (Nagai et al. 2007; Igarashi et al. 1992; Gatchel and Zoghbi 2005), as well as with aggregation properties both in vitro and in vivo (Takahashi et al. 2010; Nagai et al. 2007; Chai et al. 1999). These facts indicate that the aggregability of the mutant protein determined by the degree of expansion of the polyglutamine stretch is directly associated with the molecular pathogenesis of polyglutamine disease. It has been proposed that the expanded polyglutamine stretch undergoes conversion to a misfolded β-sheet conformation, resulting in formation of inclusion bodies through the assembly of oligomeric intermediates. Indeed, the mutated α-synuclein associated with PD also show increased aggregability (Eschbach and Danzer 2014; Lagier-Tourenne et al. 2010; Johnson et al. 2009; Austin et al. 2014), indicating the existence of a common mechanism for neurodegenerative disorders.
Another proposed mechanism of pathogenesis is one in which aggregation results from an increase in the amount of causative protein or peptide. This is analogous to the production of inclusions in hereditary neurodegenerative diseases resulting from the increasing number of genes. An increase in the number of α-synuclein genes causes familial PD (Singleton et al. 2003), and a point mutation of the gene can increase the amount of causative peptide. Moreover, increase in the number of APP genes and mutations in APP increases the production of Aβ and causes familial AD (Goate et al. 1991; Hardy and Selkoe 2002). Finally, perturbations in the proportion of messenger RNA (mRNA) splicing variants lead to production of inclusions (Goedert and Spillantini 2006). Alternative splicing of tau generates six isoforms containing either three or four microtubule-binding repeats in approximately equal proportions (Spillantini and Goedert 2013). Furthermore, mutation of MAPT, which encodes tau protein, alters the proportion of splicing variants and causes tau aggregation in several disorders (Rademakers et al. 2004; Spillantini and Goedert 2013). However, in sporadic cases the mechanism initiating the change to an aggregation-prone protein form remains unclear.
Conformational change may also occur as a secondary or tertiary disease mechanism. In AD there are two main inclusions: senile plaques and neurofibrillary tangles. A major component of the former is Aβ protein (Hardy and Selkoe 2002), and the major component of the latter is tau protein (Iqbal and Grundke-Iqbal 2006). The Aβ oligomer promotes tau phosphorylation and causes aggregation (Tomiyama et al. 2010). These facts argue for AD possibly being, primarily, a disease involving Aβ deposition and, secondarily, a tauopathy. Although tau protein is involved in neuronal cell death, it is considered from this viewpoint to have little influence on system selectivity.
1.4 The Propagation of Prion-like Protein in Neurodegenerative Disease
It has been proposed that the pathological misfolding of disease-associated proteins occurs autonomously and independently in each cell, indicating that the spread of pathological change is a consequence of these regional changes (Goedert et al. 2010; Prusiner 2012). However, a pathological feature of neurodegenerative disorders has disproved this hypothesis. Braak et al. (2003) proposed that α-synuclein inclusions in PD patients form in two regions: (1) from the dorsal vagal nucleus to the medulla oblongata and midbrain, or (2) from the olfactory bulb finally spreading to the prefrontal cortex, cingulate gyrus, and mesial temporal lobe. In order to explain the propagation of pathological misfolding of disease-associated proteins in a restricted area of the nervous system, the prion hypothesis has been proposed (Goedert et al. 2010) (Fig. 1.2A).
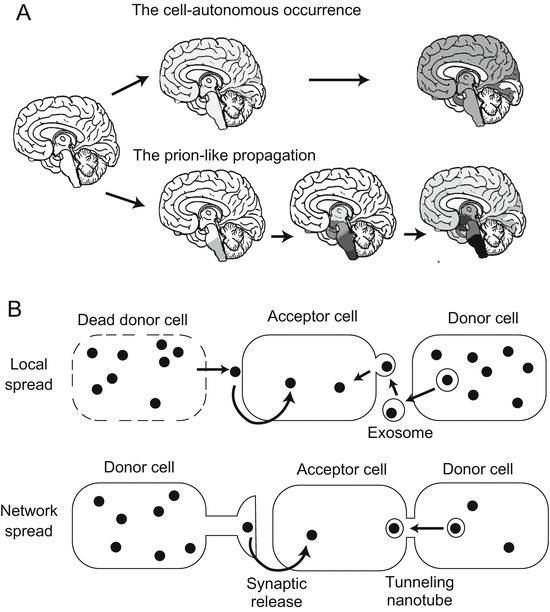
Fig. 1.2
Propagation of prion-like protein: (A) conventional supposition in which misfolded protein occurs autonomously and independently in each cell; (B) prion-like propagation theory of neurodegenerative disease, which contends that accumulation of misfolded protein begins in a specific location in the brain and progresses continuously. (This figure is based on Goedert et al. 2010 and Frost and Diamond 2010)
Prion disease is a lethal neurological disorder caused by proteinaceous infectious particles (prions) (Prusiner 1998). Normal prion protein (PrPc) is expressed systemically and is relatively highly expressed in the central nervous system (Bendheim et al. 1992). Abnormal prion protein (PrPsc), which is insoluble and rich in β-sheet structure, is derived from PrPc through a conformational change without any amino acid alteration. The PrPsc replicates itself in an autocatalytic chain reaction (Cohen et al. 1994; Lansbury 1994). The prion hypothesis expands the concept of pathologically infectious misfolded protein to other proteinopathies. This theory contends that the pathological misfolded protein is transferred to other cells, where it induces a conformational change in normally folded protein, resulting in spread of the pathological protein through the central nervous system. Indeed, transmission of pathological misfolded α-synuclein has been reported in the brain of PD patients who have received transplanted fetal neurons. α-Synuclein inclusions have been found in these transplanted fetal neurons, suggesting that pathological misfolded protein has been transmitted to them (Li et al. 2008). Moreover, transmission of misfolded protein has been proven in animal models of α-synucleinopathy (Luk et al. 2012; King et al. 2012; Li et al. 2013) and tauopathy (Clavaguera et al. 2009).
The prion-like propagation hypothesis raises the possibility that protein with a prion-like domain may cause neurodegenerative disease. The prion-like domain in RNA-binding protein has been attracting attention as a mechanism that may account for initiation of inclusion body formation in neurodegenerative diseases. King et al. (2012) have computationally predicted prion-like domains in 21,873 human proteins and actually confirmed their presence in 246 proteins (Couthouis et al. 2011). Of these 246 proteins, 49 are speculated to be RNA-binding types (Li et al. 2013), including TARDBP (King et al. 2012), FUS (King et al. 2012; Li et al. 2013), TAF15 (Couthouis et al. 2011), and EWSR2 (Couthouis et al. 2012), which are associated with neurodegenerative disease. Kim et al. (2013) have hypothesized that RNA-binding proteins with prion-like domains may cause human disease. They found pathogenic mutations of heterogeneous nuclear ribonucleoproteins (hnRNPs) A2B1 and A1 in these domains in families with inherited motor neuron disease. Moreover, causative mutations in TARDBP and FUS have frequently been observed in the prion domain. They enhance the prion-like properties of the protein (Lagier-Tourenne et al. 2010; Lattante et al. 2013). These findings lend weight to the hypothesis that prion-like propagation may underlie the molecular pathogenesis of neurodegenerative disorders.
Although accumulating evidence supports the prion-like propagation hypothesis, several issues remain to be clarified. For example, transmission has not been proven in many human neurodegenerative disorders. Transmission from tissues containing PrPsc has been demonstrated in kuru (Manuelidis et al. 2009), iatrogenic prion disease (Brown et al. 2012), and variant Creutzfeldt–Jakob disease (vCJD) in humans (Will et al. 1996). Transmission from tissue containing pathological misfolded α-synuclein and Aβ proteins in humans is still unclear (Irwin et al. 2013). Pituitary growth hormone (c-hGH) extracted from postmortem brain is known to cause iatrogenic CJD (Brown et al. 2012) with a frequency of 0.4 % (29 cases/7700 recipients) in the United States (Brown et al. 2012). On the other hand, in a follow-up study of 6190 patients out of 7700 (average age 37.3 years, mean follow-up 27.2 years), none developed AD or PD (Irwin et al. 2013). The frequency of PD and AD is significantly higher than that of CJD. Moreover, tau, Aβ, and α-synuclein can accumulate in the pituitary gland (Irwin et al. 2013). Thus, c-hGH extracted from postmortem brain might contain these misfolded proteins. These facts indicate that the infectivity of Aβ and α-synuclein is markedly lower than that of PrPsc.
Another issue is how pathological misfolded protein is transmitted to other cells. In prion disease, PrPsc spreads to the whole brain without any marked system selectivity (Parchi et al. 1999). To explain the progression of neurodegenerative disease in terms of the prion theory, it is necessary to clarify the mechanism responsible for selective propagation. Local-spread and network-spread pathways have been proposed for transcellular propagation (Frost and Diamond 2010). The former pathway is regulated by the release of aggregated proteins into the extracellular space after cell death or the release of exosomes through exocytosis (Frost and Diamond 2010; Fevrier et al. 2004). The network-spread pathway is regulated by the trans-synaptic mechanism (Frost and Diamond 2010) or through the nanotube tunnel system that links cells (Gousset et al. 2009) (Fig. 1.2B). The network-spread pathway may play an important role in explaining the system selectivity of individual neurodegenerative disorders. However, it is still unclear how such disorders stop spreading in the brain. In some cases of ALS, TDP-43 pathology is observed in the pyramidal system as well as the frontotemporal lobe, indicating that TDP-43 pathology starts in the former and then spreads to the latter. However, Nishihira et al. (2008) have reported that TDP-43 pathology in some cases of ALS is restricted to the pyramidal system, even in those showing a long clinical course. Thus, disease duration cannot explain difference in the pattern of TDP-43 pathology. Therefore, although the prion hypothesis is attractive as an explanation of the system selectivity of neurodegenerative disorders, based on our current knowledge it is still insufficient.
1.5 How Conformational Disease Causes Cell Death
Protein or peptide with a pathological conformation induce cell death or dysfunction through a mechanism that involves gain of toxicity or loss of function (Winklhofer et al. 2008). The gain-of-toxicity mechanism results from inclusion bodies acquiring a pathogenetic nature that causes dysfunction and cell death, whereas the loss-of-function mechanism involves loss of the protein’s normal function through conformational change, with resulting deleterious effects on the cell. These mechanisms are not contradictory concepts and both mechanisms might cause disease in some cases. For example, aggregation of tau inhibits normal axonal transport in tauopathy, resulting in dysfunction and cell death (gain of toxicity) (Stokin et al. 2005), whereas it also decreases the amount of normal tau, thus impairing normal function of the protein in microtubule stabilization (loss of function) (Yoshiyama et al. 2013; Weingarten et al. 1975). Although the gain-of-toxicity hypothesis holds that inclusion bodies are cytotoxic, recent studies have highlighted the importance of oligomer in the process of cell dysfunction or cell death (Takeda et al. 2006; Takahashi et al. 2010; Nagai et al. 2007; Sawada et al. 2004).
1.6 Diseases Caused by Alteration of RNA
In contrast with conformational disease, several neurodegenerative disorders are caused by alteration of RNA (O’Rourke and Swanson 2009; Cooper et al. 2009). In one such type of disease, disease-associated mutated RNAs bind to proteins and alter their function. Expansion of nucleotide repeats in the untranslated region causes neurodegenerative diseases including SCA 8, 10, 12, 31, 36, and myotonic dystrophy type 1 (Todd and Paulson 2010; de Leon and Cisneros 2008). An expansion of GGGGCC repeats in the first intron of C9orf72 has been shown to be a common cause of familial/sporadic ALS and frontotemporal lobar dementia (FTLD) (DeJesus-Hernandez et al. 2011; Konno et al. 2013). Nucleotide repeat expansion disease can show a pathogenetic mechanism that reflects both conformational disease and RNA disease: (1) the proteins that bind specifically to the unique sequence are trapped in an abnormal mRNA repeat, causing loss of protein function (Lee and Cooper 2009); (2) the mRNA repeat enhances the expression of other proteins through activation of an unknown mechanism (Lee and Cooper 2009); and (3) the nucleotide repeat in the untranslated region can express homopolymeric expansion proteins in all three reading frames, regardless of the start codon (repeat-associated non-ATG translation) (Cleary and Ranum 2013; Zu et al. 2011) (Fig. 1.3).
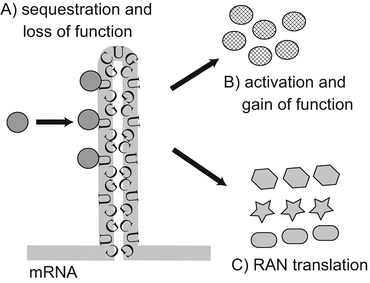
Fig. 1.3
The pathological mechanism responsible for expansion of nucleotide repeats in the untranslated region. The expanded nucleotide forms a hairpin-like structure. A CUG expansion is shown as an example: (A) RNA-binding protein is trapped specifically in the repeat expansion; (B) the repeat induces an increase in the expression level of other proteins through an unknown mechanism; (C) the repeat in the untranslated region can initiate transcription, and three forms of the protein are expressed regardless of the start codon (This figure is based on Lee and Cooper (2009))
The unknown mechanism is associated with RNA metabolism in several ways, including transcription, splicing, editing, polyadenylation, transportation, translation, and degradation of mRNA (Cooper et al. 2009). Homeostasis of RNA is termed “ribostasis” (Li et al. 2013; Ramaswami et al. 2013). We first review diseases associated with aberrant pre-mRNA splicing, which has been detected in many hereditary neurodegenerative disorders, including myotonic dystrophy type 1 and type 2 as well as fragile X-associated tremor/ataxia syndrome (FXTAS) (Licatalosi and Darnell 2006; Ling et al. 2013; Sellier et al. 2010). Most of these diseases are caused by sequestration of splicing factors in an expanded repeat motif in RNA.
1.7 Gems and Motor Neuron Disease
However, the causative protein for spinal muscular atrophy – survival of motor neuron (SMN) – has a specific function in the pre-mRNA machinery. SMN forms inclusions in the nucleus known as “Gems,” which are the sites of assembly and maturation of small nuclear ribonucleoprotein (snRNP) (Morris 1783; Coady and Lorson 2011; Dundr 2012). snRNP is an essential component of the splicing machinery (Will and Luhrmann 2005). In the assembly of snRNP, SMN first forms a dimer and binds directly to Gemin 2, 3, and 8 and indirectly to Gemin 4, 5, 6, 7, and unrip protein (Neuenkirchen et al. 2008). The SMN complex then binds to the Sm complex and uridine-rich small nuclear RNAs (U snRNAs) and transports them into the nucleus (Burghes and Beattie 2009). Gems are inclusions where additional proteins are assembled on snRNPs and U snRNAs are modified, consequently forming a spliceosome. Spliceosomes regulate the splicing of pre-mRNA and are classified as major or minor, according to consensus sequences of acceptor and donor sites for pre-mRNA splicing (Will and Luhrmann 2005). Although the major class of spliceosomes regulates most pre-mRNA splicing, minor spliceosomes also play an important role in regulating the splicing or global speed of pre-mRNA processing (Turunen et al. 2013). In spinal muscular atrophy, the number of Gems and U snRNA in the minor spliceosomes are markedly decreased, resulting in aberrant alternative splicing (Burlet et al. 1998; Gabanella et al. 2007; Shan et al. 2010; Zhang et al. 2008; Lotti et al. 2012).
TDP-43 also co-localizes with Gems (Wang et al. 2002; Tsuiji et al. 2013). The amount of TDP-43 protein affects the number of Gems (Shan et al. 2010; Ishihara et al. 2013). It has been found that Gems are decreased in affected tissue in ALS (Tsuiji et al. 2013; Ishihara et al. 2013). Furthermore, U12 snRNA, belonging to the minor spliceosome class, is decreased in tissue with TDP-43 pathology, but not without (Ishihara et al. 2013). Finally, immunohistochemical analysis has revealed that the amount of snRNP belonging to minor spliceosomes is decreased in spinal motor neurons in ALS patients (Ishihara et al. 2013). These findings are consistent with previous results obtained using an SMN-reduced mouse model (Zhang et al. 2008). However, another group has reported that an increase in U snRNAs and snRNPs subtypes is accompanied by a decrease in the number of Gems in tissues affected by ALS (Tsuiji et al. 2013). Therefore, the type of alteration of U snRNA and snRNPs occurring in ALS is still unclear.
1.8 Stress Granules and RNP Granules in Motor Neuron Disease
Several RNA-binding proteins have been identified as causative genes for motor neuron disease (Couthouis et al. 2011, 2012; Li et al. 2013; Iguchi et al. 2013). These proteins are frequently observed in cytoplasmic ribonucleoprotein (RNP) granules and stress granules (SGs), which have a role in the triage and degradation of RNA (Ramaswami et al. 2013; Kiebler and Bassell 2006). They are composed of mRNAs and RNA-binding proteins (King et al. 2012; Li et al. 2013; Ramaswami et al. 2013), suggesting that SGs play an important role in the pathogenesis of neurodegenerative disorders (Kiebler and Bassell 2006; Guil et al. 2006; Dormann et al. 2010; Anderson and Kedersha 2008). Indeed, pathological alleles of these RNA-binding proteins, such as FUS and TDP-43, facilitate the formation of SGs (Li et al. 2013; Kim et al. 2013; Wolozin 2012). Notably, disease mutations promote excess incorporation of hnRNPA2 and hnRNPA1 into stress granules and drive the formation of cytoplasmic inclusions in animal models that recapitulate human pathology (Kim et al. 2013). Thus, dysregulated polymerization caused by a potent mutant in a prion-like domain of these RNA-binding proteins can initiate degenerative disease (Kim et al. 2013). In addition, ataxin-2 increases the risk of ALS by modifying the function of SGs (Elden et al. 2010). Ataxin-2 showing intermediate expansion of the polyglutamine tract facilitates the formation of SGs, followed by inhibition of SG degradation after exposure to stress (Farg et al. 2013).
These results raise the possibility that neurodegeneration can be brought about by increasing the formation of SGs (Li et al. 2013). Several hypotheses have been proposed to explain how aggregated proteins in RNP granules inhibit normal RNA metabolism: (1) by binding to mRNA and inhibiting its expression (Nevins et al. 2003); (2) by inhibiting the protective response of SG to stress (McDonald et al. 2011; Aulas et al. 2012); and (3) by being sequestered into SG, followed by consequent loss of their own function (Aulas et al. 2012) (Fig. 1.4).
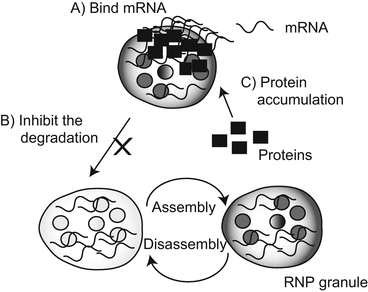
Fig. 1.4
Pathological consequences of expansion of nucleotide repeats in the untranslated region.
Several specific proteins and RNA form RNP granules, which can be synthesized and degraded rapidly in response to the state of the cell: (A) aggregated proteins bind to specific mRNA and inhibit its expression; (B) aggregated proteins inhibit the degradation and synthesis of RNP granules; (C) aggregation of the protein causes loss of its own function
1.9 Perspective
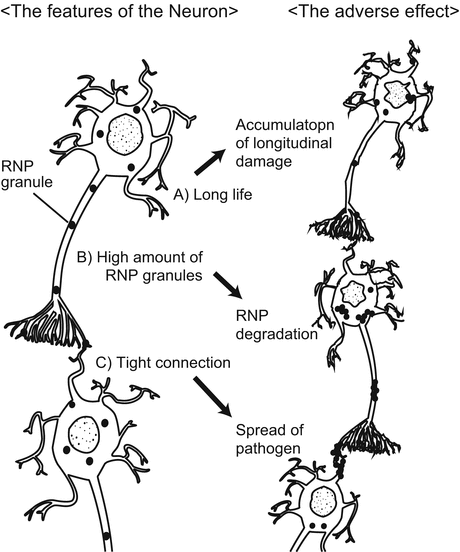
This chapter has reviewed the history of neurodegenerative disease and current concepts of the molecular pathogenesis of neurodegenerative disorders: conformational disease theory and RNA disease theory. These hypotheses are not mutually exclusive, and a combination of both might occur in several diseases (Ramaswami et al. 2013; Ling et al. 2013). Further studies of both mechanisms may lead to the development of therapeutic strategies that could theoretically include removing accumulated protein (Nicoll et al. 2003; Davtyan et al. 2013), inhibiting its accumulation (Crouch et al. 2011), regulating pre-mRNA splicing (Cooper et al. 2009), and controlling RNP granule assembly (Ramaswami et al. 2013). These mechanisms also explain the vulnerability of neurons in neurodegenerative disorders to unique features of neurons (Ramaswami et al. 2013): terminally differentiated and non-dividing cells. These features may contribute to the accumulation of unfolded proteins. In addition, neurons possess higher numbers of RNP granules in their dendrites and axons, suggesting that neurons might be vulnerable to RNP alteration. Finally, the tight connectivity of synapses may facilitate the spread of misfolded proteins or RNP granules to adjacent neurons (Ramaswami et al. 2013; Brundin et al. 2010) (Fig. 1.5).
 Parkinson’s Disease; Neurodegeneration as Systemic Disease
Parkinson’s Disease; Neurodegeneration as Systemic Disease
 Brain–Peripheral Organ Communication
Intermediate Phenotype Approach for Neuropsychiatric Disorders
Social Implementation of Neurodegenerative Disease Research and Neuroethics
Brain–Peripheral Organ Communication
Intermediate Phenotype Approach for Neuropsychiatric Disorders
Social Implementation of Neurodegenerative Disease Research and Neuroethics
 Clinical Systems Neuroscience
Clinical Systems Neuroscience
 Home- and Community-based Medical Care for Neurodegenerative Diseases: ALS as an Illustration
Home- and Community-based Medical Care for Neurodegenerative Diseases: ALS as an Illustration




Related posts:
Parkinson’s Disease; Neurodegeneration as Systemic Disease
Brain–Peripheral Organ Communication
Intermediate Phenotype Approach for Neuropsychiatric Disorders
Social Implementation of Neurodegenerative Disease Research and Neuroethics
Clinical Systems Neuroscience
Home- and Community-based Medical Care for Neurodegenerative Diseases: ALS as an Illustration
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
