Chapter 13 Congenital Cerebral Impairments
Cerebral Palsy
Preventable obstetric injuries, such as anoxia, account for less than 10% of cases. In contrast, unalterable antepartum factors account for more than 70%. For example, CP is often a manifestation of genetic or congenital malformations, such as microgyria (small cerebral gyri), pachygyria (thickened gyri), hydrocephalus, and porencephaly (see Fig. 20-4). Also, because 5% of CP children have a first-degree relative with a similar condition, as yet undetermined genetic factors undoubtedly determine or at least contribute to many cases.
Several conditions mimic CP closely enough to represent diagnostic pitfalls. Several of the disorders included in this chapter cause motor impairments that physicians may mistake for CP. In addition, insidiously advancing leukoencephalopathies (see Chapter 15) may produce spastic paresis almost identical to spastic CP. Dopa-responsive dystonia gives rise to a disorder similar to choreoathetotic CP (see later and Chapter 18). Deafness, which may occur alone or be accompanied by other neurologic disabilities, may mimic CP or mental retardation.*
Neurologists often divide CP into four varieties. Each one has a characteristic motor impairment, such as spastic paresis or choreoathetosis (Fig. 13-1), and a correlation with epilepsy and mental retardation. Neurologists usually do not diagnose CP in infants until they are at least 4 months old and, in some cases, not until they are 4 years old. Moreover, once children have an established motor deficit attributable to a perinatal cerebral injury, it must not progress as the affected child grows. In fact, impairments may seem to recede as children learn compensatory strategies and benefit from various therapies.
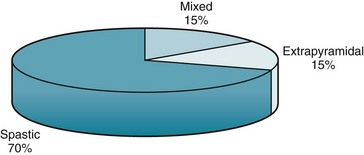
FIGURE 13-1 Most cases of cerebral palsy (CP) are varieties of spastic CP: hemiplegic, diplegic, and quadriplegic. Because studies vary, the percentages are approximations.
Although CP-induced motor impairments and associated mental retardation remain stable, comorbid epilepsy may further impair the CP child. Although epilepsy may not appear during infancy, it is usually evident before 5 years of age. Its incidence roughly corresponds to the severity of physical impairments and mental retardation (Fig. 13-2).
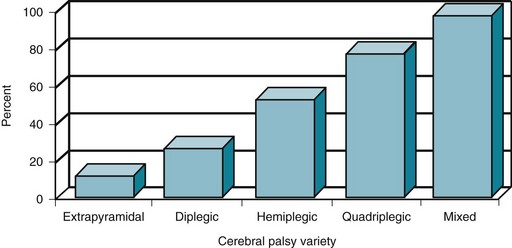
FIGURE 13-2 The proportion of cerebral palsy (CP) patients with mental retardation and epilepsy increases with more extensive cerebral disease. The incidence of those complications in choreoathetosis or extrapyramidal CP is only approximately 10%; however, the incidence in diplegic CP is 25%; hemiplegic CP 50%; quadriplegic CP 75%; and mixed CP 95%.
Spastic Cerebral Palsy
Diplegic CP (spastic diplegia) consists of bilateral symmetric paresis characteristically involving the legs more than the arms (Fig. 13-3). This CP variety usually forces children to hold their legs straight, drawn together (adducted), and crossed over each other (“scissored”). It also forces them to keep their feet and toes pointed downward (extended). When children begin to walk, this posture obligates them to stand on their toes with their legs brought closely together.
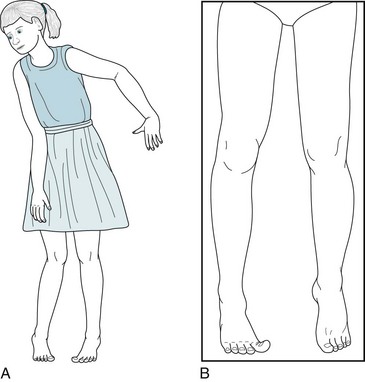
FIGURE 13-3 A, Spastic diplegia in this 10-year-old girl with low-normal intelligence causes straightening, inturning, and adduction of her legs; a tiptoe stance; and scissor-like gait. Her uncoordinated, awkward arm movements (posturing) also reflect her cerebral palsy. B, Another patient with spastic diplegia, an 18-year-old college engineering student, has typical increased muscle tone, adduction of his legs, and “toe-walking.” He also has a subtle, sustained right-sided Babinski sign.
Hemiplegic CP consists of spastic hemiparesis that typically affects the face and arm more than the leg (Fig. 13-4). The motor impairment of children and adults with hemiplegic CP resembles adults with strokes from middle cerebral artery occlusions, but they differ in three respects. While normal infants younger than 2 year old do not show hand preference, infants with hemiplegic CP show premature handedness. For example, unequivocal right-handedness in infants younger than 1 year old may mean that the left hand, if not the entire left arm, is paretic. Because left hemisphere injury during the perinatal period forces the right hemisphere to assume dominance, children and adults with congenital right hemiparesis maintain dominance in the right hemisphere and have no language impairment (aphasia). Their lack of aphasia accompanying right hemiparesis contrasts starkly with the results of a left middle cerebral artery stroke, where aphasia is often the most devastating result of damage to the mature left-sided perisylvian language arc. Finally, older children and adults with hemiplegic CP show growth arrest of the affected limbs. Compared to their normal limbs, affected ones are shorter, less muscular, and weaker.
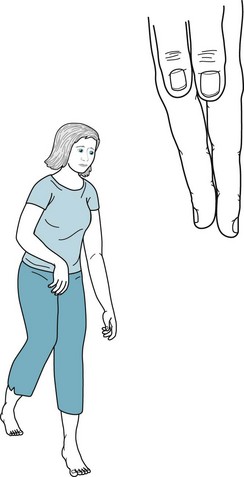
FIGURE 13-4 Hemiparetic since birth, this 28-year-old woman with normal intelligence holds her spastic and weak arm, wrist, and fingers in a flexed posture. Growth arrest of her right hand has led to shortened fingers and a less broad thumb nail bed. Similarly, her right leg, especially the heel (Achilles) tendon, is short. The growth arrest causes her to walk on her right toes and circumduct that leg. Her posture and gait are similar to that of adults after a left middle cerebral artery infarction (see Figs. 2-3 to 2-5).
As a general rule, the underlying cerebral damage in quadriplegic CP is worse than in spastic hemiparesis and much worse than in spastic diplegia. Thus, epilepsy and mental retardation occur more frequently in quadriplegic than in hemiplegic CP, and occur much more frequently than in diplegic CP. Physical and occupational therapy, bracing, and orthotics may all help these children. Neurologists often reduce spasticity by recommending surgery that transposes or lengthens tendons; prescribing oral antispasticity medications, such as baclofen and tizanidine; and administering intramuscular injections of botulinum toxin (see Chapter 18). However, epilepsy in these children resists treatment. Seizure control often requires two or more antiepileptic drugs (AEDs), which in turn may produce undesirable side effects, particularly sedation, cognitive impairments, paradoxical hyperactivity, and other behavioral disturbances.
Extrapyramidal Cerebral Palsy
Involuntary writhing movements (athetosis) of the face, tongue, hands, and feet punctuated by jerking movements (chorea) of the trunk, arms, and legs – embraced by the term choreoathetosis – define extrapyramidal CP (Fig. 13-5). Although choreoathetosis may remain subtle throughout a patient’s lifetime, it often interferes with fine hand movements, walking, and even sitting. Another manifestation – involuntary larynx, pharynx, and diaphragm movements – may lead to incomprehensible dysarthria.
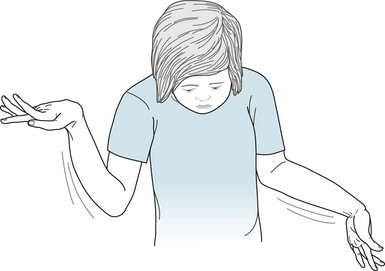
FIGURE 13-5 The slow sinuous movements (athetosis) of her wrists, hands, and fingers, present since age 3 years, signify that this 13-year-old girl has congenital cerebral palsy-induced choreoathetosis. The athetosis intermittently forces her hands into flexion at the wrist and her fingers into extension with overlapping positions.
Physicians should distinguish choreoathetotic CP from dopamine-responsive dystonia, which produces similar involuntary movements in young children (see Chapter 18). In short, unlike CP, dopamine-responsive dystonia is progressive (albeit slowly), fluctuating in a characteristic diurnal pattern at its onset, and, most important, responsive to small doses of levodopa (L-dopa). Despite the differences, the clinical similarity can be so great that many neurologists insist on a therapeutic trial of L-dopa before accepting a diagnosis of choreoathetotic CP.
Neural Tube Closure Defects
During the third and fourth weeks of gestation, dorsal ectoderm normally invaginates to form a closed, midline neural tube that eventually forms the brain and spinal cord and seals itself at both ends (Fig. 13-6, A). While ectoderm thus gives rise to the CNS as well as the skin, mesoderm forms the coverings of the CNS – the meninges, vertebrae, and skull.
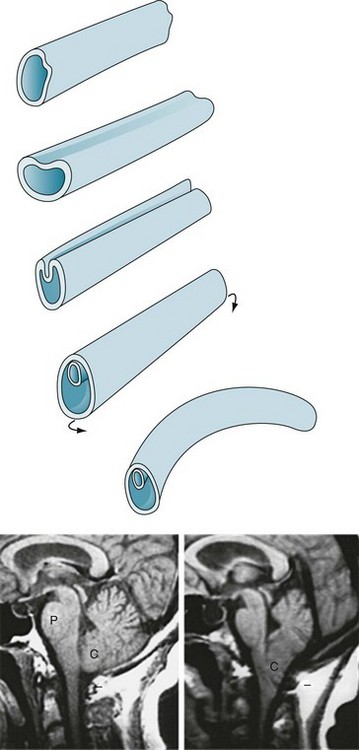
FIGURE 13-6 Top, The neural tube forms during the third and fourth weeks of gestation. Its formation begins when the embryo’s external layer, the ectoderm, invaginates to shape a distinct, midline neural tube that eventually closes at both ends. Once closed, the embryo begins to bend into a curved fetus with the tube on the convex surface. Failure to complete this process results in defects, most commonly at the upper and lower ends of the spinal cord. Bottom left, The magnetic resonance imaging (MRI) scan shows the normal relationship of several of the structures contained in the posterior fossa: the pons (P), medulla (unmarked), cerebellum (C), and the fourth ventricle (the black, cerebrospinal fluid-filled, triangular area between the pons and middle of the cerebellum). Note that the lower portion of the cerebellum remains above the foramen magnum (indicated by a short horizontal line). Bottom right, This MRI shows an Arnold–Chiari abnormality. The lower portion of the cerebellum, which includes the tonsils, and the medulla protrude below the foramen magnum. In addition, in severe cases, aqueductal stenosis causes hydrocephalus.
Upper Neural Tube Closure Defects
A group of malformations, collectively termed the Arnold–Chiari malformation, constitute a variety of upper neural tube closure defects. Usually not obvious by external appearances, the Arnold–Chiari malformation consists of downward displacement of the lower portion of the medulla and cerebellum through the foramen magnum (Figs 13-6 and 20-22). In older children and adults, who may previously have escaped detection, this malformation may produce headaches (especially when bending), bulbar palsy, and neck pain. Patients with compression of the medulla or cerebellum require “unroofing” of the upper cervical spine and occipital portion of the skull.
Lower Neural Tube Closure Defects
In meningocele, a more serious variety, the meninges and skin protrude through a lumbosacral spine defect to form a large, CSF-filled bulge. Although this condition may remain asymptomatic, it frequently causes symptoms originating in dysfunction of the lumbar and sacral nerves, such as leg weakness, gait impairment, and bladder-emptying problems. Thus progressive hydronephrosis often complicates the deficit. Meningomyelocele (myelomeningocele), which occurs far more frequently than meningocele, is the most serious variety. It consists of a tangle of a rudimentary lower spinal cord, lumbar and sacral nerve roots, and meninges protruding into a saclike structure overlying the lumbosacral spine (Fig. 13-7). The disrupted nerve tissue causes paraparesis, areflexia, and incontinence. In addition, hydrocephalus and other brain abnormalities are comorbid in about 25% of cases. Approximately 10% of infants born with meningomyelocele die from the defect.
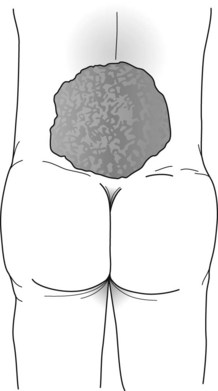
FIGURE 13-7 The meningomyelocele of this newborn infant has a typical broad-based, loose, translucent sac of thin, friable skin arising from the lumbar area. It contains rudiments of a spinal cord and lumbosacral nerves. Its surface weeps a mixture of serum and cerebrospinal fluid. The infant’s legs, lacking innervation, remain weak, flaccid, and areflexic. Similarly, the bladder, also lacking innervation, distends.
Neurocutaneous Disorders
Tuberous Sclerosis
Tuberous sclerosis usually causes conspicuous smooth and firm nodules, facial angiofibromas (adenoma sebaceum), on the malar surface of the face (Fig. 13-8), but this illness-defining skin lesion usually fails to appear until adolescence. However, during infancy and childhood, the skin shows several other characteristics: subtle hypopigmented macules (ash-leaf spots); shagreen patches, which are leathery, scaly areas, on the lower trunk and buttocks; and periungual fibromas of the fingers.
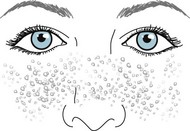
FIGURE 13-8 Adenoma sebaceum (facial angiofibromas), the cutaneous component of tuberous sclerosis, consists of nodules several millimeters in diameter, firm, and uniformly pale. They spread over the malar surface of the face. Although adenoma sebaceum may resemble acne, acne pimples have a liquid (pus) center surrounded by inflammation. Also acne pimples accumulate on the trunk as well as the face.
In another important aspect of the illness, some tuberous sclerosis children display autistic behavior. Thus, neurologists consider tuberous sclerosis as one of several neurologic causes of autism-like symptoms (Box 13-1).
Neurofibromatosis
Café-au-lait spots, the signature of neurofibromatosis, are areas of uniformly light brown, oval, and flat skin (Fig. 13-9). Although individual café-au-lait spots are found in at least 10% of normal individuals, the presence of more than six of them, each larger than 5 mm in children and 1.5 cm in adults, strongly suggests a diagnosis of neurofibromatosis. Freckling in the axilla and groin – two skin surfaces sheltered from sun exposure – often accompanies NF1-related café-au-lait spots.
FIGURE 13-9 Café-au-lait spots are flat, light-brown skin lesions. The diagnosis of neurofibromatosis requires six or more lesions, each measuring at least 1.5 cm in adults and 0.5 cm in children.
Neurofibromas consist of soft, palpable, subcutaneous growths, each a few millimeters to several centimeters in size, that emerge along peripheral nerves (Figs 13-10 and 13-11). They can also grow from nerve roots within the spinal canal and compress the spinal cord or cauda equina. They occasionally reach grotesque proportions or induce extraordinary growth of an affected limb. However, the famous nineteenth-century “elephant man,” Joseph Merrick, commonly cited as an example of neurofibromatosis, probably suffered from a related condition, Proteus syndrome.
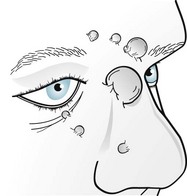
FIGURE 13-10 Neurofibromas often grow to several centimeters of disfiguring protuberances on the face.
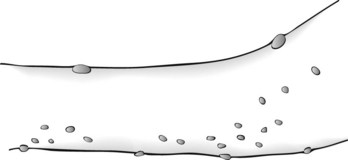
FIGURE 13-11 Neurofibromas are often subtle, multiple, subcutaneous, soft, and typically less than 0.5 cm in size.
Lisch nodules, the least obvious but most common manifestation, are multiple, asymptomatic, macroscopic, yellow to brown nodules (melanocytic hamartomas) situated on the iris (Fig. 13-12). Although a slit-lamp examination may be required to detect Lisch nodules and then differentiate them from inconsequential pigment collections, they are almost pathognomonic of the disorder.
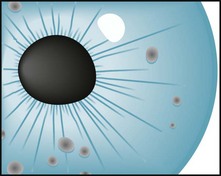
FIGURE 13-12 Lisch nodules, virtually pathognomonic of neurofibromatosis type 1, are pigmented aggregations on the iris that are often visible with the unaided eye.
Neurofibromatosis type 2 (NF2), which occurs only 10% as frequently as NF1, is an almost completely different disorder. NF2, also called familial acoustic neuroma or “central-type” neurofibromatosis, is characterized by bilateral acoustic neuromas (vestibular schwannomas) that steadily impair hearing until deafness ensues. It may induce a few neurofibromas and large, pale café-au-lait spots, but its hallmark remains the acoustic neuromas (see Fig. 20-27). In fact, NF2 is usually unrecognized until acoustic neuromas are discovered.
Sturge–Weber Syndrome
The facial angioma consists of a deep red discoloration (“port-wine stain”) in the distribution of one or more divisions of the trigeminal nerve (Fig. 13-13). Its extent does not correlate with the size of the cerebral abnormality. Clinicians must distinguish it from completely benign, more common skin abnormalities, such as small forehead angiomas (“strawberry nevi”). Also, port-wine stains, even in the trigeminal nerve distribution, are associated with Sturge–Weber syndrome in only 8% of cases. Whether or not facial angiomas are a manifestation of Sturge–Weber syndrome, laser therapy can bleach them.
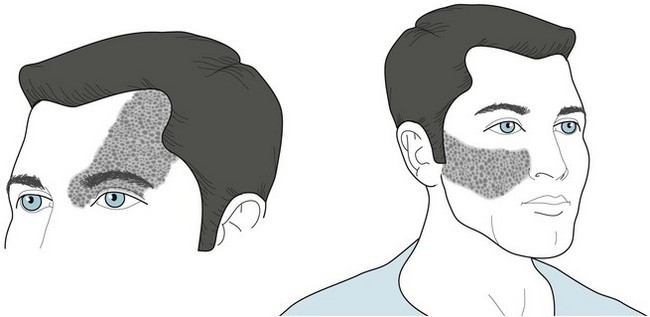
FIGURE 13-13 The cutaneous angioma (port-wine stain) of Sturge–Weber syndrome encompasses one or more divisions of the distribution of the trigeminal nerve (see Fig. 4-12). The commonest sites are the anterior scalp, forehead, and upper eyelid, i.e., the first division of the trigeminal nerve. One-third of patients have bilateral involvement.
Other Genetic Neurologic Disorders
Autosomal Chromosomal Disorders
Phenylketonuria (PKU) (Chromosome 12)
1. The deficiency would prevent the normal metabolism of phenylalanine to tyrosine. Thus, affected untreated individuals have elevated plasma concentrations of phenylalanine and little or no tyrosine.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


