, Jean Paul G. Vonsattel2, Helmut Heinsen3, 4 and Horst-Werner Korf5
(1)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University Frankfurt, Frankfurt, Germany
(2)
Medical Center Neurological Institute, Columbia University, New York, NY, USA
(3)
Division Psychiatic Clinic Morphological Brain Research Unit, Julius Maximilians University Würzburg, Würzburg, Germany
(4)
University of Sao Paulo Medical School, Sao Paulo, Brazil
(5)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University, Frankfurt, Frankfurt, Germany
5.1 Clinical Symptoms Pointing to an Involvement of the Cerebellum in Huntington’s Disease (HD)
The autosomal dominantly inherited polyglutaminopathy causing Huntington’s disease (HD) is a currently untreatable and rare neuropsychiatric disorder with an estimated prevalence in Europe and the USA of 4–8:100,000 (Andrew et al. 1993; Finkbeiner and Mitra 2008; Harper 1992; Labbadia and Morimoto 2013; Margolis and Ross 2003; Ortega et al. 2007; Rüb et al. 2009; Schapira et al. 2014; Schulte and Littleton 2011; Tanner and Goldman 1994; Vonsattel and DiFiglia 1998; Walker 2007a, b). The symptoms of adult-onset HD commonly begin around the age of 40 years with progressive cognitive impairments and motor symptoms often designated as clumsiness, tremor, balance trouble, or jerkiness. Although choreatic movements may also be among the early and progressive HD symptoms, they plateau and disappear in the advanced clinical stages. During the late clinical stage, additional somatomotor (i.e., bradykinesia, akinesia, dystonia, hypotonia, rigidity, dysarthria, dysphagia) and oculomotor symptoms, visual and executive dysfunctions, personality changes, psychiatric disturbances (e.g., depression, schizophrenia-like symptoms), electrophysiological abnormalities, and an unintended, severe, and unexplained weight loss may occur (Andrew et al. 1993; Aziz et al. 2008; Borrell-Pagès et al. 2006; Bruyn et al. 1979: Deuschl et al. 1989; Ellenberger et al. 1978; Gil and Rego 2008; Heinsen et al. 1994; Hennerici et al. 1985; Imarisio et al. 2008; Josiassen et al. 1984; Knikou 2008; Kremer et al. 1992; Lasker and Zee 1997; Leigh and Zee 2006; Li and Conforti 2013; Margolis and Ross 2003; McLeod 1969; Misiaszek 2003; Myers 2004; Petersen et al. 2005; Ross and Tabrizi 2011; Rüb et al. 2009, 2013a, 2014a, b; The Huntington’s disease Collaborative Research Group 1993; Vonsattel 2008; Vonsattel and DiFiglia 1998; Vonsattel et al. 1985; Walker 2007a, b).
The gradual worsening degeneration of the striatum has been for a long time used to explain the spectrum of HD symptoms, including oculomotor and choreatic symptoms (see Chaps. 1, 2, and 3) (Andrew et al. 1993; Borrell-Pagès et al. 2006; Braak and Braak 1992a, b; De la Monte et al. 1988; Estrada-Sanchez and Rebec 2013; Heinsen et al. 1994, 1996, 1999, Heinsen and Rüb 1997; Imarisio et al. 2008; Lange and Aulich 1986; Lange et al. 1976; Margolis and Ross 2003; Mink 1996; Myers et al. 1988; Ortega et al. 2007; Rüb et al. 2009; 2013a, 2014a, b; Vonsattel 2008; Vonsattel and DiFiglia 1998; Vonsattel et al. 1985; Walker 2007a, b). A variety of symptoms (i.e., dysarthria, impaired rapid alternating movements and fine motor skills, postural and gait instability, ataxia, falls), however, pointed to cerebellar damage in HD patients. Since the cerebellum was only fragmentarily investigated in adult HD patients, its affection during HD has been controversially discussed for several decades (Bruyn et al. 1979; Busse et al. 2009; Fennema-Notestine et al. 2004; Grimbergen et al. 2008; Jeste et al. 1984; Knikou 2008; Koller and Trimble 1985; Kremer et al. 1992; Margolis and Ross 2003; Rodda 1981; Rosas et al. 2002, 2003; Rüb et al. 2013a; Vonsattel 2008; Vonsattel and DiFiglia 1998; Vonsattel et al. 1985; Walker 2007a, b).
5.2 Neurodegeneration of the Cerebellum in Huntington’s Disease (HD)
Recently, the first systematic and comprehensive investigation of the cerebellum in clinically diagnosed and genetically confirmed HD patients clearly revealed its consistent involvement in HD patients with Vonsattel grades 2–4 of striatal atrophy (Figs. 2.11, 5.1, 5.2, 5.3, 5.4, and 5.5) (Rüb et al. 2013a). Macroscopically, the brains of the symptomatic HD patients showing extra-cerebellar symptoms (i.e., choreatic movements, bradykinesia, dysphagia, weight loss, executive dysfunctions, personality changes, cognitive decline) and/or cerebellar symptoms (i.e., unsteady and broad-based gait, gait imbalance, impaired gait coordination, dysarthria, slowed, smooth pursuit eye movements) displayed consistent atrophic changes of the cerebellum (i.e., reduction of its entire volume and of the surface area of its arbor vitae; atrophy of the lobules of the anterior and posterior lobes; widened primary fissure) (Fig. 2.11) (Rüb et al. 2013a).
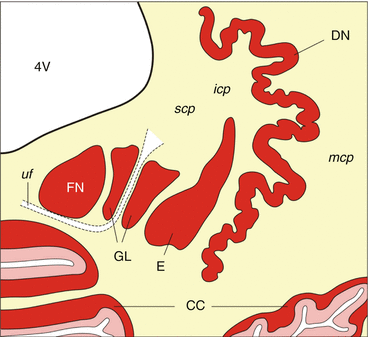
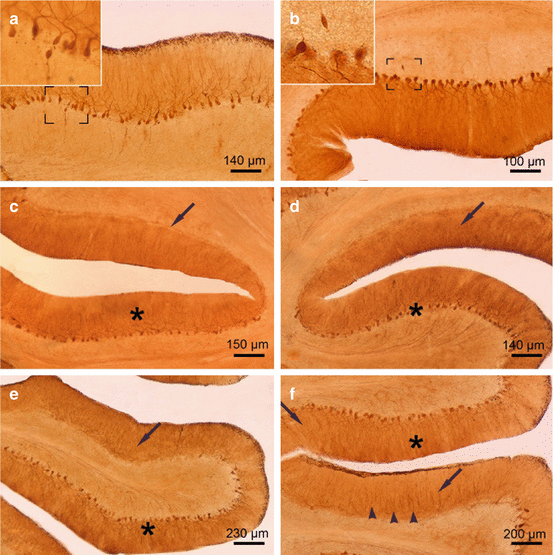
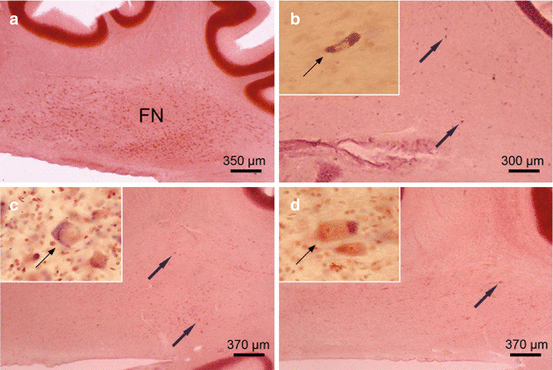
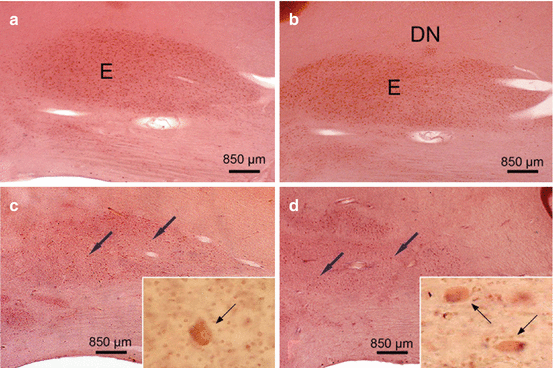
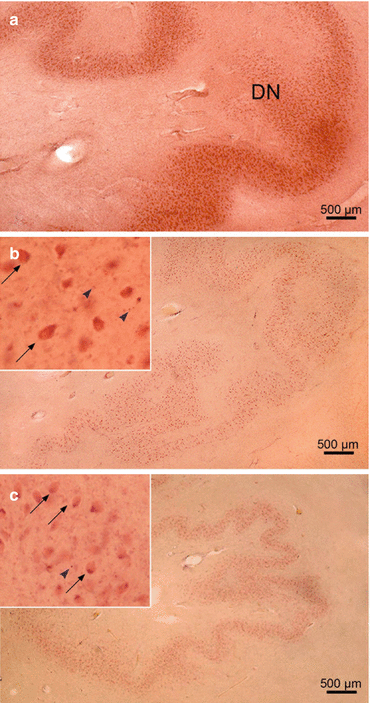
Fig. 5.1
The human cerebellum. Schematized horizontal section through the right human cerebellum showing the cerebellar cortex (CC) with its three neuronal layers (i.e., molecular, Purkinje, and granular cell layers). Embedded in the deep cerebellar white matter reside the cerebellar peduncles (icp inferior cerebellar peduncle, mcp medial cerebellar peduncle, scp superior cerebellar peduncle) and the four cerebellar nuclei (the curvated band of the DN dentate nucleus; E the cigar-shaped emboliform nucleus; GL the globose nucleus split by the uncinate fascicle (uf) into multiple portions; FN the roof ridge-like fastigial nucleus) (Modified according to Scherzed et al. (2012), (Figure 2, page 752); with kind permission from Springer Science and Business Media). Abbreviations: CC cerebellar cortex, DN dentate nucleus, E emboliform nucleus, FN fastigial nucleus, GL globose nucleus, icp inferior cerebellar peduncle, mcp medial cerebellar peduncle, scp superior cerebellar peduncle, uf uncinate fascicle, 4V fourth ventricle
Fig. 5.2
The cerebellar Purkinje cell layer in Huntington’s disease (HD). (a) Sagittal section through the vermal portion of the cerebellar lobule X of a Huntington’s disease (HD) patient with Vonsattel grade 2 of neostriatal atrophy. The Purkinje cell layer is intact and shows no loss of these cerebellar output neurons. Framed area is depicted at higher magnification at top left and shows a torpedo-like axonal inclusion. (b) Sagittal section through the vermal portion of the cerebellar lobule V of the same HD patient. Although the Purkinje cell layer is well preserved, the Purkinje cells showed torpedo-like inclusions in their axons (framed area and inset at top left). (c) Hemispheral portion of the cerebellar lobule VI of an HD patient with Vonsattel grade 4 of neostriatal atrophy: intact segments of the Purkinje cell layer (asterisk) are intermingled with sections showing complete Purkinje cell loss (arrow). (d) Hemispheral portion of lobule VI of a HD patient with Vonsattel grade 2 of neostriatal atrophy: Intact segments of the Purkinje cell layer (asterisk) and degenerated segments with serious loss of Purkinje cells (arrow). (e) Hemispheral portion of lobule VI of a HD patient with Vonsattel grade 2 of neostriatal atrophy: along with intact Purkinje cell layer segments (asterisk), portions with severe loss of Purkinje cells (arrow) prevail. (f) Hemispheral portion of cerebellar lobule IX of an HD patient with Vonsattel grade 2 of neostriatal atrophy: portions with no Purkinje cell loss, but dendritic alterations (asterisk) alternate with regions affected by severe loss of Purkinje nerve cells (arrow). Note the remaining and pathologically altered dendritic trees (arrowheads) (a–f Anti-calbindin immunostaining, 100 μm polyethylene glycol sections) (Reprinted from Rüb et al. (2013a), (Figure 2, page 168); with kind permission from John Wiley and Sons)
Fig. 5.3
The cerebellar fastigial nucleus (FN) in Huntington’s disease (HD). (a) Sagittal section through the right cerebellum of a representative control individual depicting the roof ridge-like fastigial nucleus (FN). (b) Dramatic neuronal loss in the FN of a representative Huntington’s disease (HD) patient with Vonsattel grade 2 of neostriatal atrophy. Arrows point to remaining FN nerve cells, and inset shows severely shrunken surviving FN nerve cell (thin arrow). (c) Severe neuronal loss in the FN of an HD patient with Vonsattel grade 2 of neostriatal atrophy. Remaining FN nerve cells are marked by arrows and inset shows ballooned FN nerve cell (thin arrow). These swollen nerve cells are characterized by an enlarged cytoplasm with a basophilic center, peripherally displaced nucleus, Nissl substance, and lipofuscin granules. (d) Seriously affected FN of an HD patient with Vonsattel grade 4 of neostriatal atrophy. Arrow indicates a surviving FN nerve cell, and inset shows ballooned surviving FN nerve cells (thin arrow) (a–d aldehyde-fuchsin Darrow red staining, 100 μm polyethylene glycol sections) (Reprinted from Rüb et al. (2013a), (Figure 4, page 170); with kind permission from John Wiley and Sons). Abbreviations: FN fastigial nucleus
Fig. 5.4
The cerebellar emboliform nucleus (E) in Huntington’s disease (HD). (a) Sagittal section through the right cerebellum of a representative control individual depicting the medial portion of the cigar-shaped emboliform nucleus (E). (b) Sagittal section through the right cerebellum of a typical control individual depicting the lateral portion of the cigar-shaped E and the most medial portion of the curvated band of the dentate nucleus (DN). (c) Marked neuronal loss in the medial portion of the E of a Huntington’s disease (HD) patient with Vonsattel grade 3 of neostriatal atrophy. Arrows point to remaining E nerve cells, and inset shows remaining ballooned E nerve cell (thin arrow) with bloated cytoplasm, space-consuming basophilic center, peripherally displaced nucleus, Nissl material, and lipofuscin granules. (d) Severe neuronal loss in the lateral portion of the E and marked neuronal drop-out in the most medial portion of the DN of an HD patient with Vonsattel grade 2 of neostriatal atrophy. Arrows point to remaining E nerve cells, and inset shows ballooned surviving E nerve cells (thin arrows) (a–d: aldehyde-fuchsin Darrow red staining, 100 μm polyethylene glycol sections) (Reprinted from Rüb et al. (2013a), (Figure 5, page 173); with kind permission from John Wiley and Sons). Abbreviations: DN dentate nucleus, E emboliform nucleus
Fig. 5.5
The cerebellar dentate nucleus (DN) in Huntington’s disease (HD). (a) Sagittal section through the right cerebellum of a control individual with no history of neuropsychiatric diseases depicting the folded dentate nucleus (DN). (b) Marked neuronal loss of the DN of a Huntington’s disease (HD) patient with Vonsattel grade 2 of neostriatal atrophy. Inset at top left points to surviving DN nerve cells: ballooned nerve cells with bloated cytoplasm and space-consuming basophilic center, peripherally displaced nucleus, Nissl material, and lipofuscin granules (thin arrows), and extraneuronal lipofuscin granules (arrowheads). (c) Considerable neuronal loss in the DN of an HD patient with Vonsattel grade 3 of neostriatal atrophy. Inset at top left shows remaining nerve cells: ballooned nerve cells (thin arrows), extraneuronal lipofuscin granules (arrowhead) (a–c: aldehyde-fuchsin Darrow red staining, 100 polyethylene glycol sections) (Reprinted from Rüb et al. (2013a), (Figure 4, page 174); with kind permission from John Wiley and Sons). Abbreviations: DN dentate nucleus
Upon light microscopical examination, a widespread neuronal loss was detected in the cerebellar cortex and all deep cerebellar nuclei of all HD patients studied (Figs. 5.2, 5.3, 5.4, and 5.5). Neuronal loss in the cerebellar cortex was assessed in pigment–Nissl-stained as well as in calbindin-immunostained cerebellar tissue sections and neuronal loss in the deep cerebellar nuclei in pigment-Nissl-stained tissue sections through the atrophic HD cerebella (Figs. 5.2, 5.3, 5.4, and 5.5) (Rüb et al. 2013a). Along with other proteins, calbindin is among the known calcium-binding proteins that participate in the cell signaling pathways of the second messenger calcium and represents an acknowledged, selective, and highly reliable immunocytochemical marker for the cerebellar Purkinje cells. In the cerebellum, rapid firing neurons experience high levels of calcium influx. Thus, tight calcium regulation is essential to avoid rapid neuronal death due to excessive excitatory stimulation (Bastianelli 2003). The large GABAergic Purkinje cells serve as the sole output of the cerebellar cortex, project to the deep cerebellar and brainstem nuclei, and are at 100 % immunoreactive for the calcium-binding protein calbindin (Bastianelli 2003). To highlight cerebellar Purkinje cells, calbindin immunostaining is more reliable than the traditional Nissl staining procedures, which were developed by the German neuroscientist Franz Nissl (1860–1919), since in the human cerebellum these neurons are only weakly stained by the Nissl technique (Whitney et al. 2008).
Investigation of the cerebellum of HD patients disclosed a consistent and selective loss of Purkinje cells, while the molecular and granular cell layers typically were spared (Fig. 5.2) (Rüb et al. 2013a). Loss of Purkinje cells was found in all lobules of the vermis and cerebellar hemispheres. It was not evenly distributed over the affected cerebellar lobules, but occurred at circumscribed predilection sites, which were intermingled between well-preserved and unremarkable segments of the cerebellar lobules. Surviving Purkinje cells often appeared ballooned or shrunken, and their axons in some instances harbored torpedo–like inclusions (Fig. 5.2) (Rüb et al. 2013a).
In addition to the cerebellar Purkinje cell layer, the four deep cerebellar nuclei (i.e., fastigial, globose, emboliform, and dentate nuclei) (Fig. 5.1) embedded into the cerebellar white matter also underwent a marked to severe neuronal loss during HD (Figs. 5.3, 5.4, and 5.5) (Rüb et al. 2013a). Among these deep cerebellar nuclei, the roof ridge-like fastigial nucleus commonly was most severely affected and nearly devoid of nerve cells. Most residual neurons appeared ballooned or shrunken. These swollen or ballooned fastigial nerve cells had a rounded and massively enlarged cytoplasm with a homogeneous central basophilic substance. Their Nissl substance, lipofuscin granules, and flattened nucleus were concentrated along the cytoplasmic membrane and thus mimicked central chromatolysis. By contrast, the shrunken fastigial neurons displayed a pale, slender, and arrowhead-like cytoplasm. Their central and pale nucleus was broader and stretched the abnormal cytoplasm, which was nearly devoid of Nissl substance (Fig. 5.3) (Rüb et al. 2013a).
The cigar-shaped emboliform nucleus, likewise, was consistently affected in HD patients and underwent a severe neuronal demise. As with the affected fastigial nucleus, the majority of surviving emboliform neurons were ballooned or shrunken (Fig. 5.4) (Rüb et al. 2013a). Although neuronal loss was consistently present in the globose and dentate nuclei of most of the HD patients studied, it was less severe than in their fastigial or emboliform nuclei. However, also the globose and dentate nuclei showed a clearly reduced nerve cell density and numerous surrounding lipofuscin granules in the neuropil (Fig. 5.5) (Rüb et al. 2013a). Lipofuscin granules are normal cytoplasmic constituents of nerve and glial cells and are structurally stable postmortem even after delayed or suboptimal fixation. Owing to the low affinity of glial and the high affinity of neuronal lipofuscin granules to the aldehyde-fuchsin dye, lipofuscin granules in the neuropil can serve as reliable markers of the former position of lipofuscin-laden neuronal perikarya that have disappeared from brain tissue (Braak et al. 2003b; Rüb et al. 2001, 2003a, 2004b, 2008a, b, 2009, 2013b; Scherzed et al. 2012).
Neither Purkinje cell loss nor neuronal loss in the deep cerebellar nuclei of the HD patients was correlated with the length of the CAG repeats in the mutated HD alleles, age at HD onset, duration of HD, age at death, or the Vonsattel grade of striatal atrophy (Rüb et al. 2013a).
Despite careful and systematic investigations of serial cerebellar tissue sections, no remaining nerve cells in the cerebellar cortex and deep cerebellar nuclei were found to display morphological features (i.e., chromatin condensation, nuclear fragmentation, apoptotic bodies) associated with the occurrence of classical apoptosis (Graeber and Moran 2002; Rüb et al. 2013a).
Reactive astrocytes immunopositive for the glial fibrillary acidic protein (GFAP) could be observed in all cerebellar components of HD patients. They were most frequent in the cerebellar white matter, but also consistently present in the three layers of the cerebellar cortex, as well as in all four deep cerebellar nuclei (Rüb et al. 2013a).
5.3 The Functional Relevance of Cerebellar Affection in Huntington’s Disease (HD)
Since the cerebellum was only fragmentarily investigated in adult HD patients, its involvement during HD has been controversially discussed for several decades (see Chap. 1) (Bruyn et al. 1979; Fennema-Notestine et al. 2004; Jeste et al. 1984; Koller and Trimble 1985; Kremer et al. 1992; Margolis and Ross 2003; Rodda 1981; Rosas et al. 2003; Rüb et al. 2013a; Vonsattel 2008; Vonsattel and DiFiglia 1998; Vonsattel et al. 1985; Walker 2007a, b). This situation has changed since the first comprehensive study of the cerebellum was published (Rüb et al. 2013a). This recent study clearly demonstrated consistent pathological changes involving the cerebellum of HD patients with Vonsattel grade 2–4 striatal atrophy. It also showed unequivocally that cerebellar neurodegeneration in HD is not confined to the Purkinje cell layer and/or the dentate nucleus as assumed previously by some authors, but includes macroscopically visible cerebellar atrophy, widespread loss of Purkinje cells, as well as a considerable and consistent demise of nerve cells in all four deep cerebellar nuclei (i.e., fastigial, globose, emboliform, and dentate nucleus) (Figs. 2.9, 5.1, 5.2, 5.3, 5.4, and 5.5) (Bruyn et al. 1979; Jeste et al. 1984; Rodda 1981; Rosas et al. 2003; Rüb et al. 2013a; Vonsattel and DiFiglia 1998; Vonsattel et al. 1985). These new and consistent neuropathological findings conform to the results from recent in vivo magnetic resonance imaging (MRI) studies (Fennema-Notestine et al. 2004; Rosas et al. 2003; Rüb et al. 2013a) and are highly reminiscent to the cerebellar findings in autosomal dominantly inherited polyglutamine ataxias (e.g., spinocerebellar ataxias types 1, 2, and 3). All findings indicate that the brain neuropathology of the polyglutamine disease HD is more closely related to these polyglutamine ataxic disorders than previously thought and underscore the contemporary concept of HD as a multisystem degenerative disease affecting the striatum, cerebral neo-and allocortex, thalamus, brainstem, and cerebellum (see Chaps. 1, 2, and 3) (Atkin and Paulson 2014; Borrell-Pagès et al. 2006; Braak and Braak 1992a, b; Bruyn et al. 1979; de la Monte et al. 1988; Dom et al. 1976; Dunlap 1927; Estrada-Sanchez and Rebec 2013; Fennema-Notestine et al. 2004; Ferrante et al. 1987; Finkbeiner and Mitra 2008; Hedreen et al. 1991; Heinsen et al. 1992, 1994, 1996, 1999; Heinsen and Rüb 1997; Hoche et al. 2008; Imarisio et al. 2008; Jeste et al. 1984; Koller and Trimble 1985; Kremer et al. 1992; Lange 1981; Lange et al. 76; Lastres-Becker et al. 2008; Margolis and Ross 2003; McCaughey 1961; Myers et al. 1988; Rodda 1981; Rosas et al. 2003; Rüb et al. 2008a, 2013a, b; Scherzed et al. 2012; Schulte and Littleton 2011; Seidel et al. 2012; Sieradzan and Mann 2001; Sotrel et al. 1991; Valera et al. 2005; Vogt and Vogt 1920, 1942; Vonsattel 2008; Vonsattel and DiFiglia 1998; Vonsattel et al. 1985; Walker 2007a, b).
Related posts:
 Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
 Elucidation of the Role of the Premotor Oculomotor Brainstem Nuclei in the Pathogenesis of Oculomotor Dysfunctions in Huntington’s Disease (HD)
Elucidation of the Role of the Premotor Oculomotor Brainstem Nuclei in the Pathogenesis of Oculomotor Dysfunctions in Huntington’s Disease (HD)
 Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
 Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
 Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


