Control of Neuronal Excitability
Uwe Heinemann
Istvan Mody
Yoel Yaari
Introduction
The behavior of nerve cells is determined by ionic channels. These are transmembrane proteins with a pore that permits ions to pass across the lipophilic membrane. Ion channels may be permanently open or regulated by stretch, voltage, or chemical compounds. Currents flow through these channels in an inward or outward direction dependent on the driving force, which is set by the difference between the actual membrane potential and the equilibrium potential for the given ion(s). An inward current depolarizes the membrane and is carried either by movement of positive charges (Na+, Ca2+) into the cell or of negative charges (Cl–) out of the cell. Conversely, outward currents drive the membrane potential in a hyperpolarizing direction and are carried either by cation (K+) currents out of or anion (Cl–) currents into the cell. At resting membrane potential, which is negative in all cells, inward and outward currents balance each other. The proteins that encode the water-filled, voltage-dependent ion channels are called α subunits. They are often associated with β, γ, and other auxiliary subunits. These may modulate properties of the channels, but they also regulate membrane anchoring and trafficking of ion channels. The receptor-gated ion channels usually consist of different α, β, and so on subunits, which all are required to form the ion channel. The nomenclature for ion channels is rather diverse. Physiologists and biophysicists originally named most ion channels according to their ion selectivity and dependence on voltage, ligand binding, or role in cellular function such as stretch, generation of receptor potentials, and so on. A second nomenclature often derives from studies of mutations in the fruit fly Drosophila. Scientists who originally cloned ion channels developed yet another nomenclature, and, finally, human geneticists use yet another. This makes reading the original literature difficult and sometimes time-consuming. I find the International Union of Basic and Clinical Pharmacology (IUPHAR) compendia on ion channels rather useful in this respect because it gives all the names used in the literature for all voltage-gated ion channels. I will follow the 2005 nomenclature of ion channels proposed by the International Union of Pharmacological Sciences. The most comprehensive survey of properties of ionic currents and their physiologic meaning is given by Hille.65
Receptor-gated ionic channels mediate the information traffic between cells and set the membrane potential as a function of changes in the intracellular milieu such as those of adenosine triphosphate (ATP), pH, or calcium. Voltage-gated ionic channels determine how a neuron integrates synaptic information and propagates it to another neuron or to effector organs. Voltage-gated channels regulate the membrane potential, influence the integrating properties of the dendrites and the discharge mode of a cell, and are responsible for the generation and propagation of action potentials. At the presynaptic terminals, they influence Ca2+ loading of the terminals, which is a prerequisite for transmitter release. It is important to note that the distribution of ion channels over the surface of a neuron is not homogeneous. Neurons are polarized cells, which often express in their dendrites other channels than in the soma, the axon, and the presynaptic terminals. Regulation of sorting and direction into axonal and dendritic transport in neurons is much less understood than in epithelial and endothelial cells, which are also polarized. The anchoring of ion channels at distinct sites within and outside the synapse is another important issue because it determines the strength of synaptic coupling and the integration properties of neurons. This has become clear because mutations in stargazing and other anchoring proteins can contribute to epileptogenesis.102 Similarly, proteins involved in vesicle cycling and other presynaptic functions can contribute to epileptogenesis.5
For ionic currents to flow, an electrochemical gradient must be provided, which depends on transport processes across the neuronal membrane. Changes in membrane potentials can be brought about by electrogenic active or secondarily active transport, and such processes frequently influence the responsiveness of neurons. Therefore, in this chapter, I first consider the operation of such ion transport mechanisms. Subsequently, I treat the properties of channels that control the resting membrane potential. I then describe voltage-operated ionic channels that account for neuronal excitability and different discharge modes of neurons. The excitability of neurons is also controlled by low-affinity subsynaptic γ-aminobutyric acid (GABA), glycine, acetylcholine, and glutamate ionotropic receptors and by high-affinity ionotropic or metabotropic receptors, which are often located at extrasynaptic sites.
Properties of Ion Transporters and Generation of Membrane Potentials
A simple way for ions to cross the plasma membrane is by means of energy-driven pumps, which use the energy from ATP to overcome the barrier imposed by the plasma membrane. Ion pumps are proteins responsible for generating and maintaining the concentration gradients of Na+, K+, Ca2+, H+, and Cl– ions across the plasma membrane; of H+ across vesicular membranes; and of Ca2+ across the mitochondrial membrane and the endoplasmic reticulum. They bind ions on one side of the membrane, physically transport it across the bilayer, and release it on the other side. Because energy is expended in this process (ATP hydrolysis), it is possible for such active transporters to move ions against a concentration gradient. The energy for this transport comes directly from ATP. It is therefore important that the respiratory chain is intact. The Na,K-ATPase is formed
by α and β subunits. Four α subunits and four β subunits have been identified, and two α and β subunits are necessary for the transport process.55,145 The ATP-dependent ion transporters affect the electrical behavior of a cell in two ways: They set up the electrochemical ionic gradients that underlie current flow when voltage- or ligand-gated channels are activated. They also affect membrane potential because the transporters are often electrogenic. As an example, Na,K-ATPase transports three Na+ ions out of a cell in exchange for only two K+ ions. Hence, the Na,K-ATPase imposes a hyperpolarizing drive on the membrane potential and thereby drives the membrane potential in a negative direction. In fact, the reversal potential for this transporter is quite negative. The membrane potential of neurons and glia would sit very much negative to -100 mV; if not, conductances for Na+, K+, and Cl– would clamp the membrane potential to more depolarized potentials. In fact, it is the Na-K pump that causes K+ to accumulate within cells and sets up the gradient for Na+ to enter cells once Na+-permeable channels are open.133 Because under resting conditions mostly K+ channels are open, K+ ions tend to leave the cells along their chemical gradient. This is prevented by the retaining force set up by negatively charged proteins. Thus, an equilibrium between influx and outflux of K+ is generated. This equilibrium is described by the Nernst equation. A membrane potential dominated by the K+ concentration gradient over the membrane would be close to -90 mV. This situation is approached in many astrocytes,116 which therefore respond to a change in extracellular K+ concentration more effectively than do neurons.
by α and β subunits. Four α subunits and four β subunits have been identified, and two α and β subunits are necessary for the transport process.55,145 The ATP-dependent ion transporters affect the electrical behavior of a cell in two ways: They set up the electrochemical ionic gradients that underlie current flow when voltage- or ligand-gated channels are activated. They also affect membrane potential because the transporters are often electrogenic. As an example, Na,K-ATPase transports three Na+ ions out of a cell in exchange for only two K+ ions. Hence, the Na,K-ATPase imposes a hyperpolarizing drive on the membrane potential and thereby drives the membrane potential in a negative direction. In fact, the reversal potential for this transporter is quite negative. The membrane potential of neurons and glia would sit very much negative to -100 mV; if not, conductances for Na+, K+, and Cl– would clamp the membrane potential to more depolarized potentials. In fact, it is the Na-K pump that causes K+ to accumulate within cells and sets up the gradient for Na+ to enter cells once Na+-permeable channels are open.133 Because under resting conditions mostly K+ channels are open, K+ ions tend to leave the cells along their chemical gradient. This is prevented by the retaining force set up by negatively charged proteins. Thus, an equilibrium between influx and outflux of K+ is generated. This equilibrium is described by the Nernst equation. A membrane potential dominated by the K+ concentration gradient over the membrane would be close to -90 mV. This situation is approached in many astrocytes,116 which therefore respond to a change in extracellular K+ concentration more effectively than do neurons.
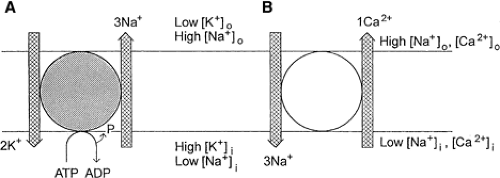 FIGURE 1. A: Active transporter. Principles of action of the electrogenic Na+,K+-ATPase. One adenosine triphosphate (ATP) molecule is used to transport three Na+ ions against the concentration gradient out of the cell, and two K+ ions are transported against the concentration gradient into the cell. B: Secondarily active transporter. In the example of this antiporter, three Na+ ions are moved along the electrochemical gradient into the cell, and one Ca2+ ion is moved against the electrochemical gradient out of the cells. Note that all transporters have a reversal potential. ADP, adenosine diphosphate. |
Most neurons and muscle cells have membrane potentials more positive than -90 mV. This is because channels permeable to Na+ and Cl– are open under resting conditions. These conductances are much smaller than those for K+. Nevertheless, the existence of Na+ leak channels implies that for each K+ ion that leaves a nerve cell, one Na+ ion can enter. The cells would slowly depolarize to zero if the Na-K pump did not become activated on intracellular accumulation of Na+ and thus restore the concentration gradients. Thus, the Na-K pump ultimately is responsible for the generation of the Na+ and K+ concentration gradients and the membrane potential. The Na-K pump can be activated by accumulation of both extracellular K+ and intracellular Na+.58 During a seizure resulting from activation of Na+ and K+ channels, Na+ accumulates within the neurons and K+ in the extracellular space. This leads to activation of the electrogenic Na+ pump and then to a hyperpolarizing drive. This hyperpolarizing drive contributes to the termination of seizures and is responsible for the long-lasting afterhyperpolarization that follows a single seizure.61 When Na+ pumps lose their efficacy, afterhyperpolarizations become smaller, and such loss of efficacy may well underlie episodes of status epilepticus. Indeed, it has been shown that accumulation of intracellular Ca2+ can impair the function of the pump.50 This may be caused by depolarization of mitochondrial membranes, which interferes with the generation of ATP,89 by consumption of ATP in pumping Ca2+ out of cells and into intracellular stores, or by a direct modulation of Na,K-ATPase.
The electrochemical gradient set up by such pumps can be exploited by secondarily active transporters. These use the electrochemical force for inward or outward movement set up by active pumps to transport a second molecule against the concentration gradient. Such transporters import glucose, amino acids, and other agents into the cells as well as export metabolites, calcium, and protons. Often these transporters are not electrically neutral, and thus they can influence the membrane potential (FIGURE 1).
Secondarily active pumps are also involved in regulating the intracellular and extracellular ionic environment. They can use the electrochemical gradient for one ion species to move another ion in the same direction (cotransport) or in the opposite direction over the membrane (antiport). Such transporters do not affect the membrane potential if they are electrically neutral. However, in many cases the transporter is not electrically neutral. For example, the Na+-Ca2+ exchanger in most cases transports three Na+ ions into the cell against one Ca2+ ion out of the cell, thus providing a depolarizing drive to the cell.13,14
Secondarily active transporters have a reversal potential. Thus, when the membrane potential moves beyond the reversal potential, the transport direction is also reversed. For example, the reversal potential for the Na+-Ca2+ exchanger is somewhere around -30 mV. This implies that depolarizations beyond this potential would drive Ca2+ into the cell, and there is indirect evidence that this occurs during spreading depression and anoxic depolarization.84 Because the transmembrane ionic gradients strongly change during such conditions, however, the reversal potential also shifts. It is therefore necessary to consider in any of these conditions the actual reversal potential for a given transporter. This also applies to situations in which GABA or glutamate is released by reversed transport from nonvesicular compartments.2,3
The intracellular Cl concentration is also affected by secondarily active transporters and in addition by Cl channels. Early during neuronal development, the NKCC1 transporter sets the Cl equilibrium potential above the resting membrane potential of neurons.119 As a result, inhibitory transmitters such as GABA and glycine produce depolarizations that early during development are sufficient to excite neurons.28 These GABA-driven giant depolarizing potentials are part of early spontaneous depolarization waves in most neuronal structures.1 Later in life (in humans, likely during the last trimester of embryonic development83), the NKCC1 transporter is replaced by the KCC2 transporter, which extrudes Cl from cells and sets the reversal potential of Cl to values below the resting membrane potential, probably dependent on certain trophic factors.82 Even then, however, the direction of transport is regulated by the extracellular potassium concentration, and it may reverse
transport direction at about 5.5 mM, resulting in a depolarizing Cl equilibrium potential.76
transport direction at about 5.5 mM, resulting in a depolarizing Cl equilibrium potential.76
Ion Channels Determining Resting Membrane Potentials
The K channels that most strongly contribute to generation of resting membrane potentials are likely two-pore domain potassium channels consisting of four transmembrane loops with two pores.57 The K2P channels are encoded by KCNK genes, with 11 members so far of the family expressed in the brain. These channels can be influenced by stretch, protons, oxygen tension, and lipids (e.g., arachidonic acid, anadamides), but also by volatile anesthetics and drugs such as riluzole, quinine, quinidine, and bupivacaine, as well as barium at concentrations of 1 to 2 mM.92 K2P channels are outwardly rectifying and are likely a major route by which potassium is released from cells when they depolarize.
The resting membrane potential is also influenced by potassium inward rectifying (Kir) channels. Seven families of these channels are encoded by KCNJ genes.90 These consist usually of four subunits with two transmembrane segments. These channels rectify because Mg and polyamines block the outflow of potassium through them. Kir channels frequently increase conductance when K accumulates outside the cells. Strongly rectifying Kir 3 family members are activated by G proteins through βγ subunits and weakly rectifying Kir 6 channels by the ratio of ATP to adenosine diphosphate (ADP). Kir 2 channels are expressed apart from neurons on muscle cells and are involved in vasodilation when K accumulates outside smooth muscle cells.29 Kir 4.1 channels (perhaps together with Kir 5.1) form the astrocytic K channels involved in spatial K buffering.74 These channels are very sensitive to Ba in low concentrations. This is in contrast to Kir 7 family members, which are only blocked by Ba at high concentration of about 100 μM. Studies on weaver mice suffering from epilepsy75 have clarified that mutations in the selectivity filter in the pore-forming loop lead to permeability also for sodium ions and resulting depolarization when Kir 3.3 channels are activated by G proteins.
The ion channels that confer Na and Ca permeability to the membrane at resting membrane potential are not well understood. Some transient receptor potential (TRP) channels may be involved.30,115 These are rather unselective cation channels with little rectification, which are thought to be involved in receptor potential generation. However, some of them are widely distributed throughout the brain. Thus, all TRPC member channels are expressed in the brain. They are usually activated by the Gq type of G proteins, diacylglycerol (DAG), and also neurotrophic factors. TRPV1–4 channels are expressed not only in peripheral, but also in many central neurons. TRPM2–4 channels are also expressed in brain and are modulated by arachidonic acid, redox state, and cytokines.
Ion channels that likely also influence membrane resting potential are the cyclic nucleotide–regulated ion channels.69 They are rather unselective cation channels permeable for both sodium and potassium. They play an important role in phototransduction, but many of them are also expressed in central neurons. They are regulated by intracellular nucleotides such as guanosine 3′,5′-cyclic monophosphate (cGMP) or cyclic adenosine 3′,5′-monophosphate (cAMP) and show little voltage dependence. This is in contrast to the hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels, which are also relatively unselective cation channels. These ion channels are activated by hyperpolarization and shift the membrane potential back toward resting membrane potential. The activation curves for these ion channels are regulated by cAMP and cGMP. There are four subunits of these ion channels, all of which are expressed in the brain. They are frequently involved in membrane potential oscillations and are also important for rhythmogenesis in thalamic neurons and other neurons displaying membrane potential oscillations. Cells that express these channels often show resonance properties: This implies that neurons become especially sensitive to specific frequencies of synaptic input usually in the θ and α frequency ranges. Upregulation of such channels may be involved in homeostatic plasticity but also following hyperthermia-induced convulsions.26
Which chloride channels are contributing to resting membrane potential is unclear. Two calcium-dependent chloride channels have been cloned from rat brain, but their functional role is unclear. Whether-volume activated chloride channels are expressed in neurons is unclear, but such channels could be involved in osmoregulation of astrocytes.
Voltage-Gated Ionic Channels
These channels determine the excitability of neurons, the different firing modes, the integrating properties of dendrites, and the mechanisms that lead to transmitter release from presynaptic terminals. Except for voltage-dependent chloride channels, most voltage-gated ion channels belong to one superfamily.160 They all build on a pore-forming unit contained in the α subunits. Voltage-regulated Na and Ca channels consist of four homologous motifs with six transmembrane α helices termed S1 to S6 with a membrane reentrant loop between S5 and S6. The voltage-gated potassium channels consist of four α subunits, which combine to form a voltage-regulated potassium channel. As in voltage-gated Na and Ca channels, the ion-conducting pore and selectivity filter are formed by the S5 and S6 segments and the reentrant pore loop between them. Four subunits are also used to form cyclic nucleotide–gated (CNG), HCN, and TRP channels. Heteromeric assembly of different subunits, combinations with different auxiliary subunits, RNA editing, and splicing confer very different properties to these channels.
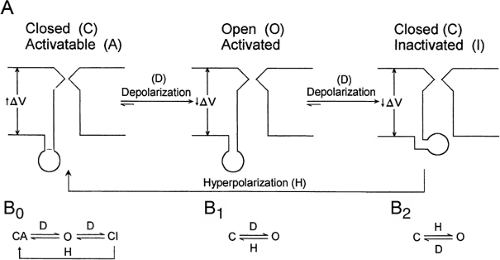 FIGURE 2. Reaction schemes of voltage-gated channels. A: Principle of action of transient Na+ or K+ channels. At rest, the channel is closed by the position of a gate in the outer mouth of the channel (closed, activatable). On depolarization, the channel proteins change their conformation, permitting passage of ions. The conformation change is controlled by a voltage sensor that measures the potential difference across the neuronal membrane. On further depolarization, an inner, ball-like structure moves into the inner mouth of the channel, obstructing the passage of ions (closed, inactivated). Hyperpolarization is required to change the conformation from the inactivated state to the activatable state. B0, B1, B2: Summary of typical reaction schemes of voltage-operated channels. C, closed; CA, closed activatable; CI, closed inactivatable; D, depolarization; H, hyperpolarization; O, open. |
Voltage-gated ionic channels are thus membrane-spanning proteins that form a pore; the opening and often also the closing of the pore are regulated by the transmembrane voltage gradient. The voltage sensor is contained in the S1-to-S4 segment of the α subunits, forming the channel, which is absent in Kir and K2P channels. Voltage-gated ionic channels usually have auxiliary subunits.160 The voltage-gated sodium channels possess only four β subunits, which modulate channel activation and regulate membrane surface expression. Therefore not only mutations in α subunits but also those in β subunits can lead to epilepsy. Voltage-gated Ca channels are regulated by β, γ, and α2δ subunits. The four β subunits are all intracellular proteins that regulate channel gating and surface expression. Mutations in β subunits can thus also lead to channelopathies, including epilepsy. There are eight γ subunits including stargazing,102 which link the Ca channels to other transmembrane proteins, including α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA)-type glutamate receptors. The α2δ subunits are binding sites for anticonvulsants such as pregabalin and gabapentin.39
Voltage-regulated K channels have different types of auxiliary subunits.93 KV1 channels are associated with β1–3 subunits; KV4 channels with Kchip 1–4; and KV3, 4, 7, 10, and 11 channels with the five members of mink-like subunits.160 The β subunits in the KV1 channel family influence channel closing. Kchips belong to the superfamily of Ca sensor proteins and regulate channel kinetics and channel membrane surface expression. The mink-like subunits are also important regulators
of channel dynamics. Finally, the KV2 family is modulated by KV5, 6, 8, and 9 modifier/silencer subunits.
of channel dynamics. Finally, the KV2 family is modulated by KV5, 6, 8, and 9 modifier/silencer subunits.
The gating of these ionic channels occurs preferentially at a defined membrane potential at which the probability of channel opening increases strongly. However, because the sensor for the gating is located within the membrane, it recognizes the voltage difference between the inside and outside leaflets of the membrane. On both sides of the membrane, there are negative surface charges that attract preferentially Ca, Mg, and H ions. Consequently, changes in H+, Ca2+, and Mg2+ and proton concentration can influence the gating properties.66 Occasionally, these ions can also modify permeability properties. Well-known examples are cation channels, which can become activated during rapid decreases in Ca2+ concentration.63,156
In addition, ionic channels can be influenced by intracellular metabolism. For example, certain K channels are rather sensitive to the ATP content of a cell. At physiologic ATP levels, these channels are closed, whereas depletion of ATP leads to opening of K channels and consequently to hyperpolarization. Block of such channels can induce seizures.7 Some ionic channels possess redox sites such as voltage-gated K channels and K2P channels, which react to the formation of free oxygen radicals or are sensitive to arachidonic acid and its derivatives.103 Most ionic channels change their properties on phosphorylation, and many protein kinases and some phosphatases can therefore affect the opening and closing of voltage-gated channels. In some cases, such modulations give the cells distinctly different discharge modes, whereas in others, there is a continuum of modifications in the reactivity of a given neuron. In addition, the intracellular level of Ca2+ can cause activation and occasionally inactivation of ionic channels.
Based on permeability of the pore, the resulting transmembrane currents are differentiated as Na+, K+, Ca2+, and Cl– currents. Proton currents have also been described and are a prerequisite for formation of radical oxygen species released from activated microglial cells.43 Within these different species of ionic currents, a distinction is made between persistent and transient currents. Most of the persistent channels open on depolarization and some on hyperpolarization of the membrane potential with respect to the resting membrane potential. Such channels behave according to the reaction schemes illustrated in FIGURE 2B. When persistent ionic currents are activated, the channels open and close, with an increased probability of being open for the whole period of time that the membrane potential is above the threshold for channel opening.
Transient currents show a time-dependent variation of probability of being open when the membrane potential is above that required for channel gating. Their reaction scheme can be formally described as in FIGURE 2A. In these channels, the open probability strongly decreases with time. This time-dependent inactivation is often mediated by a ball formed by the intracellular end of transmembrane proteins. These balls move into the inner mouth of the channels, thereby preventing further passage of ions. This type of inactivation is designated chain-and-ball inactivation and resides at the N terminal of the channel protein.66 By contrast, there is also a C-type inactivation, which depends on conformational changes in the channel itself. Inactivation of either type makes the channel temporarily refractory and induces a time-dependent short-term memory into channel properties. The conditions and time it takes to remove inactivation are usually studied in paired pulse experiments. Removal of inactivation in sodium channels is typically delayed when anticonvulsants such as carbamazepine or phenytoin are applied.118 Besides the kinetics of an ion channel, the steady-state behavior also is used to describe the channel properties.68 Steady-state inactivation curves are determined by current measurements with pulses evoked from different membrane potentials to a potential at which the current is maximal. Conductances are then derived from Ohm’s law by taking into account the driving force for the ion species and the measured current amplitude. Normalized conductance curves are then plotted and give the percentage of ion channels that can be maximally opened as a function of resting membrane potential. The steady-state activation curve describes the relative conductance as a function of the membrane potential achieved during a voltage step. A leftward shift of the steady-state
inactivation curve then indicates that at a given membrane potential a fewer channels are available for current generation. Such leftward shifts of steady-state inactivation curves contribute to effects of anesthetics and anticonvulsants. A leftward shift of the steady-state activation curve indicates that more channels can contribute to an ionic current and for the sodium channel would indicate a lower threshold for the generation of action potentials.
inactivation curve then indicates that at a given membrane potential a fewer channels are available for current generation. Such leftward shifts of steady-state inactivation curves contribute to effects of anesthetics and anticonvulsants. A leftward shift of the steady-state activation curve indicates that more channels can contribute to an ionic current and for the sodium channel would indicate a lower threshold for the generation of action potentials.
Thus, voltage-gated ionic channels can frequently be modulated by drugs. These often affect membrane fluidity, changing the energy barriers that a channel protein must overcome to change its conformation from a closed to an open state. Moreover, drugs can bind to channel proteins, stabilizing a given conformation and changing its threshold for activation. Especially interesting are drugs that are use dependent, that is, they bind to a channel protein only when it is activated. Such drugs often enter the open mouth of an ionic channel and then move very slowly through the channel, preventing further current flow. In some cases, they affect the transition from an inactivated to an activatable state and thereby also produce a use-dependent effect. On the other hand, some drugs and toxins impede the inactivation process, changing a transient into a persistent current.
Voltage-Gated Sodium Currents
The fast transient Na+ currents are mainly involved in the generation of action potentials, first described by Hodgkin and Huxley.67 The rapid upstroke of the action potential is the result of a voltage-dependent increase in membrane Na+ conductance. The threshold for action potential generation is defined as the membrane potential at which Na (and Ca) conductances equal those of K (and Cl) conductances. This implies that Na channels must also be active in the subthreshold range. In most central neurons, the threshold of activation for the fast Na+ current is about -60 mV.23 The current–voltage relationship of this current is characteristically a U-shaped curve, for which the maximum of current amplitude is usually reached at about -20 mV. The fast transient Na+ currents inactivate rapidly. The inactivation behavior of the current has been described with time constants of 2 to 4 msec in most neurons studied. The Na channel can only reactivate when inactivation is removed. This is a voltage-dependent and time-dependent process often involving two time constants.118 Rapid removal of inactivation is typical for interneurons, which therefore can fire in higher frequencies than most glutamatergic neurons. By means of electrophysiologic and biochemical techniques, several toxins have been identified as blocking Na+ channels. After it was found that tetrodotoxin (TTX) acts in nanomolar concentrations as a selective blocker of most neuronal Na+ channels, it became a useful chemical tool for studying properties of these channels in more detail.80 In addition to TTX-sensitive channels, TTX-resistant Na+ channels have been detected.159 Acutely isolated neurons from the medial entorhinal cortex of the rat exhibit a TTX-resistant Na+ current in addition to the TTX-sensitive Na+ current.154 Like TTX, saxitoxin and μ-conotoxin are able to block Na+ conductance by either occluding the channels or causing conformational changes.31 Moreover, a number of lipid-soluble alkaloid toxins, including batrachotoxin, veratridine, aconitine, and grayanotoxin, can shift the voltage dependence of activation in a hyperpolarizing direction and prevent current inactivation, resulting in persistent channel activation at normal membrane potentials.24 The voltage dependence of Na+ currents is also changed by the application of β-scorpion toxins, which cause enhanced activation. Brevetoxins and ciguatoxins can cause repetitive firing of nerve cells by shifting the activation curve of Na+ currents to more negative potentials.24 Toxins such as veratridine and brachatoxin can therefore readily induce convulsant activity.107
Sodium currents are encoded by nine different α subunits, which combine with four different β subunits. NaV1.1–1.3, 1.5, and 1.6 are expressed in central neurons. NaV1.1 (SCN1A) channels associate with β1 to β4 subunits.24 NaV1.1 channels are highly expressed in hippocampal interneurons, and deletion of this channel leads to reduced firing of interneurons in the hippocampus, which explains some of the hereditary epilepsies associated with mutations in these channels such as generalized epilepsy with febrile seizures plus (GEFS+) and myoclonic epilepsy. The β4 subunit in this and other channels accelerates removal of inactivation, permitting high-frequency generation of action potentials, as is typical for many interneurons. NaV1.2 (SCN2A) can also interact with all β subunits. It is highly expressed in axons of central neurons. Mutations in these channels have also been linked to epilepsy.143 NaV1.3 channels are highly expressed during development but are also expressed on somata in adult brain cells. These channels are upregulated after nerve injury and, due to rapid removal of inactivation, can be involved in high-frequency axonal discharges. This implies that the action potential frequency in somatic recordings may not necessarily be the same in axon terminals. They interact with β1 and β3 subunits. NaV1.5 channels (SCN5A) are insensitive to TTX and, besides heart muscle cells, are also expressed on subgroups of central neurons. They can interact with all four β subunits. Finally, NaV1.6 (SCN8A) channels are expressed in central neurons and mostly on the somatodendritic region of output cells in the cerebral cortex, hippocampus, and cerebellum. It is thought that these ion channels contribute at least partially to persistent sodium channels.
The β subunit 1 occurs with different mutations, leading to GEFS+ and also to temporal lobe epilepsy (TLE).73 Persistent noninactivating Na+ current has been found in a number of neurons.4,6,48 Its activation threshold was determined at potentials more negative than that of the fast transient Na+ current, at about -70 mV. Because of activation near the resting membrane potential of neurons and the loss of inactivation, this current can contribute to subthreshold membrane potential oscillations and resonance, playing an important role in the genesis of the θ rhythm in these cells.4 It is likely that also some metabolites can remove inactivation and thereby permit generation of persistent sodium currents. Another possibility is that persistent sodium channels are due to window currents. When the steady-state activation and inactivation curves are considered, they often overlap and generate a voltage range in which a portion of these channels is frequently open.
Voltage-Gated Calcium Currents
Calcium currents play a major role in neuronal excitability, directly by their contribution to membrane depolarization and indirectly through the elevation of the intracellular concentration of free Ca2+. Voltage-dependent Ca2+ conductances in neurons contribute to the generation of dendritic spikes, slow somatic depolarizations, and related burst discharges.22 Influx of Ca2+ through voltage-gated Ca2+ channels can activate Ca2+-dependent K+ channels and regulate many intracellular Ca2+-dependent processes. Calcium channels are also responsible for rapid delivery of Ca2+ to trigger transmitter release.46,150
Voltage-gated Ca2+ channels consist of α1 subunits with which β subunits, α2δ, and γ subunits can be associated.25 In the classic studies of sensory neurons by Carbone and Lux,19 they showed that voltage-gated Ca2+ channels could be differentiated into two major categories: (a) low-voltage-activated (LVA) and (b) high-voltage-activated (HVA)
channel types. The α1 subunits of Ca channels can be differentiated into three families: (a) CaV1, (b) CaV2, and (c) CaV3 members.25 The CaV1 family shares high threshold of activation, no voltage-dependent inactivation, and sensitivity to dihydropyridines, and these are often referred to as L-type Ca channels. In the central nervous system (CNS), only Ca V 1.3 (α1c, CACNA1C) channels are expressed on somata and dendrites of neurons, with little effect on regulation of transmitter release. The CaV2 subfamily (CaV2.1–2.3) has three members, which are all insensitive to dihydropyridines. CaV2.1 (α1A, CACNA1A, or P/Q-type) channels are involved in transmitter release but are also found in many neurons expressed on dendrites. They are sensitive to ω-agatoxin IVa. The CaV2.2 (α1B, CACNA1B, or N type) channels are also expressed presynaptically and on dendrites. These channels show time-dependent inactivation and are blocked by ω-conotoxin GVIA. Like CaV1 channel members, these two channels require high levels of depolarization to become activated. CaV 2.3 channels (α1E, CACNA1E, or R-type channels) require less strong depolarization for activation, are expressed on somata and dendrites, and are sensitive to the peptide SNX-482. They can be involved in epilepsy.144 Low-voltage-activated channels contain CaV3.1 to 3.3 members (α1G, H, I; CACNA1G, H, I), are rather sensitive to low concentrations of Ni, and form the T-type Ca channels. In most neurons, these channels activate at potentials positive to -70 mV. Inactivation of the T-type Ca2+ channels develops monoexponentially within tens of milliseconds and shows a strict dependence on voltage. Unlike in L-type Ca channels, the inactivation kinetics is independent of Ca2+ influx through the channel.20,21 No specific high-affinity antagonist has so far been identified for the T-type Ca2+ current, although antiepileptic drugs used to treat petit mal, such as ethosuximide and dimethadione, reduce T-type Ca2+ currents in thalamic neurons and dorsal root ganglion cells.35
channel types. The α1 subunits of Ca channels can be differentiated into three families: (a) CaV1, (b) CaV2, and (c) CaV3 members.25 The CaV1 family shares high threshold of activation, no voltage-dependent inactivation, and sensitivity to dihydropyridines, and these are often referred to as L-type Ca channels. In the central nervous system (CNS), only Ca V 1.3 (α1c, CACNA1C) channels are expressed on somata and dendrites of neurons, with little effect on regulation of transmitter release. The CaV2 subfamily (CaV2.1–2.3) has three members, which are all insensitive to dihydropyridines. CaV2.1 (α1A, CACNA1A, or P/Q-type) channels are involved in transmitter release but are also found in many neurons expressed on dendrites. They are sensitive to ω-agatoxin IVa. The CaV2.2 (α1B, CACNA1B, or N type) channels are also expressed presynaptically and on dendrites. These channels show time-dependent inactivation and are blocked by ω-conotoxin GVIA. Like CaV1 channel members, these two channels require high levels of depolarization to become activated. CaV 2.3 channels (α1E, CACNA1E, or R-type channels) require less strong depolarization for activation, are expressed on somata and dendrites, and are sensitive to the peptide SNX-482. They can be involved in epilepsy.144 Low-voltage-activated channels contain CaV3.1 to 3.3 members (α1G, H, I; CACNA1G, H, I), are rather sensitive to low concentrations of Ni, and form the T-type Ca channels. In most neurons, these channels activate at potentials positive to -70 mV. Inactivation of the T-type Ca2+ channels develops monoexponentially within tens of milliseconds and shows a strict dependence on voltage. Unlike in L-type Ca channels, the inactivation kinetics is independent of Ca2+ influx through the channel.20,21 No specific high-affinity antagonist has so far been identified for the T-type Ca2+ current, although antiepileptic drugs used to treat petit mal, such as ethosuximide and dimethadione, reduce T-type Ca2+ currents in thalamic neurons and dorsal root ganglion cells.35
The behavior of voltage-gated Ca currents is influenced by different subunits. The intracellular β subunit consists of four known isoforms β1 to β4 (CACNB1–CACNB4). Mutation in β4 subunits also are involved in ataxia and epilepsy in mice and humans.18 The β subunits regulate current density by controlling the amount of α1 subunit expressed at the cell membrane. In addition to this trafficking role, the β subunits regulate the activation and inactivation kinetics and shift the voltage dependence for activation of the α1 subunit pore in the hyperpolarizing direction.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


