Chapter 8 Cortical Myoclonus and Epilepsy: Overlap and Differences
Introduction
The term myoclonus is used to describe a brief and jerky involuntary movement involving antagonist muscles and originating from brief active contractions of muscles (positive myoclonus) or, more rarely, from brief interruptions of ongoing electromyographic activity (negative myoclonus).1 Clinically, myoclonus may be classified as “focal” if it involves a restricted, usually distal, group of muscles; “multifocal” when asynchronous focal jerks involve different body areas; or “generalized” when jerks involve most body segments in an apparently synchronous manner.
Myoclonus is spontaneous if occurring irrespective from external triggering situations or can be induced by movement (action myoclonus) or by sensory or visual stimuli (reflex myoclonus). Finally, regarding periodicity, myoclonus may be either rhythmic or arrhythmic.
Many conditions in which myoclonus is a prominent manifestation are known, allowing an etiological classification1,2 in which four major clinical syndrome categories are identified: (1) physiologic myoclonus (sleep-related, hiccup, myoclonus induced by anxiety or exercise); (2) essential myoclonus (individuals without other neurological signs); (3) epileptic myoclonus (conditions in which the predominant element is epilepsy); and (4) symptomatic myoclonus (conditions in which the predominant element is encephalopathy).
Neurophysiological characteristics can be used to divide myoclonus into six main physiological categories, including cortical, cortical-subcortical, subcortical-supraspinal, spinal, and peripheral.3
The term epileptic myoclonus is still confusing. Some authors define as epileptic myoclonus what occurs within the setting of epilepsy.4 Others define as epileptic myoclonus those forms in which a paroxysmal depolarization shift is thought to be the underlying neurophysiological substrate, irrespective of which population of neurons (cortical or subcortical) is primarily involved.5 In our opinion, epileptic myoclonus can be comprehensively defined as an elementary electroclinical manifestation of epilepsy involving descending neurons, whose spatial (spread) or temporal (self-sustained repetition) amplification can trigger overt epileptic activity.6
Frequently, the electroencephalogram (EEG) correlate of epileptic myoclonus can be detected only by using jerk-locked (EEG or magnetoencephalogram) averaging. Yet, many patients with cortical myoclonus have rhythmic electromyogram (EMG) bursts at relatively high frequency (especially those with minipolymyoclonus, cortical tremor, Angelman syndrome or autosomal dominant cortical myoclonus, and epilepsy), which make it difficult to identify a cortical correlate. Recent works have demonstrated in these cases the role of EEG-EMG coherence by means of frequency analysis in demonstrating common cortical drives.7,8,9
Myoclonus can be only one component of a seizure (myoclonic jerks heralding a generalized tonic-clonic seizure in juvenile myoclonic epilepsy or in progressive myoclonus epilepsies), the only seizure manifestations (myoclonic jerks of benign myoclonic epilepsy), one of multiple seizure types (as observed in myoclonic-astatic epilepsy), or the basis of a movement-related disorder (action myoclonus in progressive myoclonus epilepsies). However, the relationships between myoclonus and epilepsy have been elucidated only in part, and the neurophysiological bases, nosology, and electroclinical characteristics of myoclonus in the setting of specific epilepsy syndromes are only partially understood. According to available evidence, epileptic myoclonus can be classified neurophysiologically as cortical (positive and negative), secondarily generalized, thalamocortical and reticular reflex myoclonus.10 Cortical epileptic myoclonus constitutes a fragment of partial or symptomatic generalized epilepsy; thalamocortical epileptic myoclonus is a fragment of idiopathic generalized epilepsy.5 Reflex reticular myoclonus, which does not have a time-locked EEG correlate, represents the clinical counterpart of hypersynchronous activity of neurons in the brainstem reticular formation. In the following sections, major epilepsy or neurological syndromes featuring different forms of epileptic myoclonus are described.
Cortical Myoclonus (CM)
CM originates from abnormal neuronal discharges in the sensorimotor cortex. Abnormally firing motoneurons may be primarily hyperexcitable or may be driven by abnormal inputs originating from hyperexcitable parietal11 or occipital12 neurons. Each jerk originates from the discharge of a small group of cortical motoneurons, somatotopically connected to a group of contiguous muscles. A cortical potential, time locked to the myoclonic potential and localized on the contralateral sensorimotor region, can be demonstrated by EEG, magnetoencephalogram, or jerk-locked averaging.13–16 Facilitation of interhemispheric and intrahemispheric spread of CM activity through transcallosal or corticocortical pathways seems to play a major role in producing generalized or bilateral myoclonus.17
In patients with cortical reflex myoclonus (CRM), appropriate stimuli administered to a resting somatic segment produce a reflex muscle response (jerk) with a latency of around 50 msec (C-reflex).18 A similar response is only observed in normal subjects during voluntary contraction. Somatosensory evoked potentials (SEPs) of giant amplitude are typically seen in association with CRM.19–22 The striking resemblance in latency and morphology of the giant SEPs to the myoclonus-related cortical spikes suggests that both originate from common cortical mechanisms.15
In the typical forms of CRM, the reflex jerk in the hand has a latency of ∼50 msec, and the CRT has a mean duration of about 7 msec.23 Typical CRM can be observed in patients with focal cortical lesions,18 spinocerebellar degeneration,14,24,25 multiple system atrophy,25,26,27 cerebral anoxia,14,28 childhood metabolic degenerations such as neuronal ceroid lipofuscinosis and sialidosis,11,19 Alzheimer’s disease,29,30 Down syndrome, and mitochondrial disorders.31,32,33
Epileptic negative myoclonus (ENM) is characterized by brief (50 to 400 msec) muscle inhibitions with focal, multifocal, or bilateral distribution and time locked to sharp-wave or spike-wave discharges on the contralateral central areas.34,35 ENM has a wide etiological spectrum ranging from idiopathic to symptomatic forms due to cortical dysplastic lesions.36 It may occasionally be precipitated by an adverse reaction to antiepileptic drugs.37,38,39 Previous studies35,40,41 hypothesized a cortical origin of ENM. Epileptic activity associated with ENM was described in the premotor42,43,44 and postcentral somatosensory cortex.40,45 Through cortical electrical stimulation studies it was suggested that negative motor areas might be present in the lateral and mesial portion of frontal lobe, encompassed in the supplementary sensorimotor area (SMA).46 However, no subsequent confirmatory studies are available.
A possible role of the SMA in ENM was also proposed in a recent study47 in which electrical stimulation of the SMA constantly evoked ENM, with no preceding positive myoclonus, as it was instead observed following stimulation of the premotor, primary motor, and sensorimotor cortex.
EPILEPTIC SYNDROMES AND NEUROLOGICAL DISORDERS WITH CM
Cortical Action/Reflex Myoclonus
Progressive Myoclonus Epilepsies (PMEs)
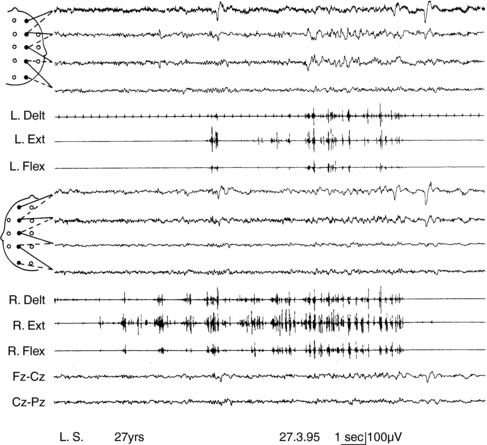
Progressive myoclonus epilepsies represent a clinically and etiologically heterogeneous group of diseases with a progressive course, characterized by myoclonus, generalized, tonic-clonic seizures, and neurological deterioration.48 Onset is most frequently in late childhood or adolescence.49 Different forms are known, including Unverricht-Lundborg disease, Lafora disease, neuronal ceroid-lipofuscinosis, type III Gaucher disease, infantile and juvenile GM2-gangliosidosis, some mitochondrial encephalopathies, sialidosis, dentatorubropallidoluysian atrophy, and action myoclonus-renal failure (AMRF).49,50 Causative genes have been identified for most PMEs.51,52 Onset features comprise myoclonus and rare generalized tonic-clonic seizures, as in idiopathic myoclonic epilepsies.53,54 The tonic-clonic seizures can occur without any warning or after a long buildup of myoclonic jerks. The EEG shows generalized polyspike and spike-and-wave discharges, frequently precipitated by photic stimulation. Background EEG activity becomes progressively slower.53 Cortical reflex myoclonus is common to all PME syndromes, in which it is manifested with the classic combination of action myoclonus (Figure 8-1), spontaneous jerks, giant SEPs, C-reflex at rest, and the premyoclonus spike. According to Cantello et al.,55 focal subcortical reflex myoclonus can also be demonstrated in these patients. Initially mild, myoclonus becomes increasingly disabling during the course. Severe action myoclonus has a devastating impact on the patients’ level of autonomy.
Rett Syndrome
Rett syndrome is an X-linked dominant disorder, with an estimated prevalence of 1 in 10,000 to 15,000 females, making it one of the most common causes of severe mental retardation in females. Mutations in the exons 1 to 4 of the methyl CpG binding protein 2 gene (MECP2)56 have been identified in roughly 75 to 80% of girls with classical Rett syndrome;57 in MECP2-negative patients additional screening using (multiplex ligation probe amplification) MLPA enables detection of large deletions in nearly half of the remaining patients with the syndrome.58 The clinical phenotype includes progressive cognitive deterioration leading to dementia, autistic features, truncal ataxia/apraxia, loss of purposeful hand movements, breathing abnormalities, stereotypies, extrapyramidal signs, and epilepsy. A form of CRM characterized by prolonged C-reflex (65 ± 5 msec) latency has been described in affected girls.59 Myoclonus is multifocal and arrhythmic, and major myoclonic seizures are not seen in these patients. A positive potential, localized on the contralateral centroparietal area, precedes myoclonus with a latency of 34 ± 7 msec for the forearm muscle compatible with corticomotoneuronal conduction. The N20-P30 and P30-N35 components of the SEPs have significantly increased amplitude. In addition, the latency of the N20 component is delayed, and the N20-P30-N35 interval is significantly increased and has expanded morphology. It is probable that in Rett syndrome the following sequence of events occurs: slight delay in central conduction of the impulse afferent to the sensorimotor cortex (N20), slowing of the processing of the afferent impulse (interval N20-P30; mean = 11 msec), delay in corticocortical transmission to the precentral neurons subserving movement of the stimulated body segment (latency increase P30 – C reflex; mean = 32 msec), and rapid descending volley to the spinal motoneurons. Intracortical conduction time could be particularly prolonged because of the synaptic abnormalities that have been observed.60
Huntington’s Disease
In Huntington’s disease, action myoclonus is a rare manifestation, but a few patients have been described in whom CRM was the presenting symptom.33,61 Seizures are an infrequent complication and are mainly seen with juvenile onset, rarely presenting with a typical PME syndrome.62
Postanoxic Encephalopathy
Postanoxic encephalopathy is characterized by dysarthria, ataxia, pyramidal signs, rigidity, epilepsy, and myoclonus, which is usually spontaneous and action-induced, multifocal and generalized, and extremely disabling. Electromyographic silent periods following the jerks contribute to producing postural lapses.63 Postanoxic myoclonus may be cortical in origin, involving the sensorimotor cortex and rapidly conducting pyramidal pathways.14,28 More rarely it may have brainstem origin, either as an exaggerated startle reflex or as reticular reflex myoclonus.64,65 Forty percent of patients with postanoxic myoclonus suffer from generalized epileptic seizures.
Focal Cortical Repetitive Myoclonus
Epilepsia Partialis Continua
Epilepsia partialis continua, or Kojewnikow’s syndrome,66 is characterized by almost continuous, focal, rhythmic (around 1 to 2 Hz) muscle jerks, which are observed both while awake and asleep, for periods ranging from hours to days, or rarely years.48 Unilateral somatomotor seizures are constantly associated. Two types of epilepsia partialis continua have been identified.67 The first type is due to fixed epileptogenic lesions involving the motor cortex. Causative factors include ischemia, posttraumatic head injury, cortical dysplasia, tumors, and vascular malformations.67–70 A stable motor deficit, predating seizure onset, and nonprogressive evolution are usual features. A second type of epilepsia partialis continua is observed in Rasmussen’s syndrome. Onset occurs during childhood, with continuous focal jerking and intractable homolateral motor or generalized seizures. Progressive hemiparesis, hemianopia, and eventually, cognitive deterioration follow. Magnetic resonance imaging (MRI) shows progressive atrophy of the affected hemisphere. Pathological studies reveal inflammation with perivascular infiltrates and microglia nodules.71 A viral etiology was originally hypothesized. More recently, the role of antibody-mediated mechanisms and more recently cell-mediated immunity have been hypothesized72,73,74 with inconclusive results. An analogous form of progressive epilepsia partialis continua has been observed in some children with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS).75
Rhythmic High-Frequency Cortical Myoclonus (Cortical Tremor)
Cortical tremor is a form of rhythmic myoclonus, presenting as postural or action tremor in some patients with progressive myoclonus epilepsy (PME),76,77 in Angelman syndrome, and in different forms of autosomal dominant epilepsy.78–83 The nosologic boundaries between epilepsia partialis continua and this peculiar form of repetitive myoclonus are unclear.
Angelman Syndrome
Angelman syndrome is a neurogenetic disorder deriving from a defect in maternal chromosome 15q11 to q13. Seventy percent of patients present a cytogenetic or molecular deletion encompassing three subunits of receptor α for gamma-aminobutyric acid (GABRB3, GABRA5, and GABRG3) and the gene UBE3A. Uniparental paternal disomy for chromosome 15, or mutations in the imprinting center or in the UBE3A gene, are more rarely found. Patients have microbrachycephaly, severe to moderate mental retardation, absence of speech, inappropriate paroxysmal laughter, epilepsy, ataxic gait, tremor, and jerky movements. Neurophysiologic investigations reveal a spectrum of manifestations of myoclonus.84 All patients present with rapid distal jerking of fluctuating amplitude, which causes a sort of coarse distal tremor combined with dystonic limb posturing. Jerks occur at rest in prolonged runs. In addition, the majority of patients have myoclonic and absence seizures, as well as episodes of myoclonic status. Bilateral jerks of myoclonic absences show rhythmic repetition at ∼ 2.5 Hz and are time locked with a cortical spike. Interside latency of both spikes and jerks is consistent with transcallosal spread, and spike-to-jerk latency indicates propagation through rapid conduction corticospinal pathway. A contralateral, central premyoclonic potential is uncovered by jerk-locked averaging. SEPs are normal, and C-reflex is absent.
Familial Adult Myoclonic Epilepsy and Autosomal Dominant Cortical Reflex Myoclonus and Epilepsy
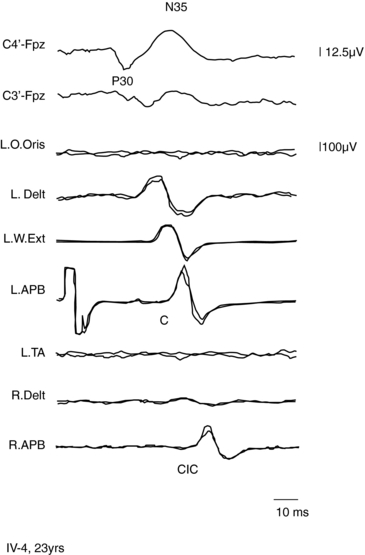
A form of autosomal dominant epilepsy with cortical myoclonus manifested as cortical tremor has been described in several families, mostly of Japanese origin,79,85 and given the acronym BFAME (benign familial adult myoclonic epilepsy) or FAME (familial adult myoclonic epilepsy). Affected patients present homogeneous characteristics, including (a) autosomal dominant inheritance; (b) adult onset (mean age 38 years, range: 19 to 73); (c) nonprogressive course; (d) distal, rhythmic myoclonus enhanced during posture maintenance (cortical tremor); (e) rare, apparently generalized, seizures often preceded by worsening of myoclonus; (f) absence of other neurological signs; (g) generalized interictal spike-and-wave discharges; (h) photoparoxysmal response; (i) giant SEPs and hyperexcitability of the C-reflex (Figure 8-2); and (j) cortical EEG potential time locked to the jerks. The original Japanese families linked to chromosome 8q23.3-q24.86 However, European families with a similar phenotype did not link to the same locus.85,87,88
Autosomal dominant cortical reflex myoclonus and epilepsy (ADCME)81 has been described in patients with a homogeneous syndromic core, including an association of nonprogressive cortical reflex myoclonus, manifested as semicontinuous rhythmic distal jerking (cortical tremor), generalized tonic-clonic convulsions (GTCs) preceded in some patients by generalized myoclonic jerks, and generalized EEG abnormalities. Age at onset of cortical tremor and of GTCs overlapped in a given individual but varied between individuals, ranging from 12 to 50 years. This clinical picture shares some features with FAME86; however, all ADCME patients had in addition focal frontotemporal EEG abnormalities, and some individuals also had focal seizures, of variable severity, starting around the same age as the other manifestations. The pattern of inheritance is autosomal dominant with high penetrance. Linkage analysis identified a critical region in chromosome 2p11.1-q12.2.81 Three Italian families with familial adult myoclonic epilepsy have been described, possibly linked to 2p11.1-q12.2, suggesting a possible allelism with ADCME.88,89
Recently two novel families with adult myoclonic epilepsy have been reported,82,83 neither linking to known loci. In the first report, clinical photosensitivity was a prominent feature; in the second report photosensitivity was not a prominent feature, but cerebellar ataxia, dementia, and progression of symptoms were observed, raising doubts about the nosology of this disorder.
Epileptic Syndromes with Secondarily Generalized Epileptic Myoclonus
Severe Myoclonic Epilepsy in Infancy (SMEI), or Dravet Syndrome
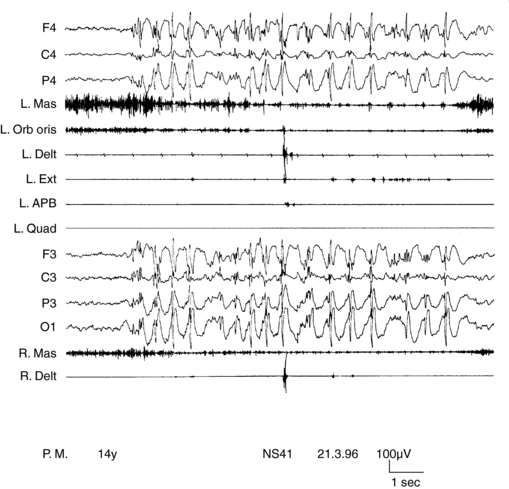
SMEI, or Dravet syndrome, is observed in 6 to 7% of children with seizure onset in the first year of life90 and is a severe form of epilepsy, characterized by multiple seizure types and unfavorable prognostic outlook. Its classification as a form of myoclonic epilepsy is controversial because myoclonus, although present in most children, can be a transient phenomenon and often does not represent a hallmark of the syndrome. A subgroup of children does not exhibit myoclonic seizures at all.90,91 SMEI represents the prototype of an epileptic encephalopathy in which onset of severe, prolonged seizures precedes deterioration of cerebral functions.92 Mutations of the SCN1A gene are observed in about 80% of cases.93 Onset of epilepsy occurs during the first year of life with prolonged generalized or unilateral, clonic or tonic-clonic seizures during fever, often evolving to status. They rapidly become associated with similar nonfebrile attacks. By the third to fourth year of life, resistant myoclonic, partial seizures and atypical absences also appear. EEG, normal at the beginning, subsequently shows multifocal and generalized abnormalities. Early photosensitivity is seen in some children. Neurological development appears delayed from the second year of life onward. Two main types of myoclonus have been described. Almost all children show arrhythmic, distal jerks, manifested as twitching of fingers, whereas some also have generalized jerks. Demonstrating a premyoclonic potential for multifocal jerks may be difficult, even using jerk-locked averaging. Generalized jerks have an obvious EEG correlate, which appear to originate from spread of CM activity, when small time differences are measured (Figure 8-3).10,54
< div class='tao-gold-member'>
Related posts:
 The Life-Threatening Epilepsies of Childhood and Their Treatment
The Life-Threatening Epilepsies of Childhood and Their Treatment
 The Spectrum of Epilepsies Associated with Generalized Spike-and-Wave Patterns
The Spectrum of Epilepsies Associated with Generalized Spike-and-Wave Patterns
 Epilepsies Due to Monogenic Disorders of Metabolism
Epilepsies Due to Monogenic Disorders of Metabolism
 The Surgery of Temporal Lobe Epilepsy I—Historical Development, Patient Selection, and Seizure Outcome
The Surgery of Temporal Lobe Epilepsy I—Historical Development, Patient Selection, and Seizure Outcome
 Mechanisms of Action of Levetiracetam and Newer SV2A Ligands
Mechanisms of Action of Levetiracetam and Newer SV2A Ligands
 Does Early Treatment Influence the Long-Term Outcome of Epilepsy?
Does Early Treatment Influence the Long-Term Outcome of Epilepsy?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


