Chapter 4 Cranial Nerve Impairments
Individually, in pairs, or in groups, the cranial nerves are vulnerable to numerous conditions. Moreover, when a nerve seems impaired, the underlying problem might not be damage to the cranial nerve itself but rather a cerebral injury, neuromuscular junction problem, or psychogenic disturbance. Following custom, this chapter reviews the 12 cranial nerves according to their Roman numeral designations, which readers may recall with the classic mnemonic device, “On old Olympus’ towering top, a Finn and German viewed some hops” (Box 4-1).
Olfactory (First)
From the olfactory receptors located deep in the nasal cavity, branches of the pair of olfactory nerves pass upward through the multiple holes in the cribriform plate of the skull to several areas of the brain. Some branches terminate on the undersurface of the frontal cortex, cornerstone of the cortical olfactory sensory area. Others terminate deep in the hypothalamus and amygdala – cornerstones of the limbic system (see Fig. 16-5). The olfactory nerves’ input into the limbic system, at least in part, accounts for the influence of smell on psychosexual behavior and memory. Also notable is that smell is the only sensation that innervates the cerebral cortex and deeper structures without intervening synapses in the thalamus or its extension, the geniculate bodies.
Although mundane problems underlie most cases of bilateral anosmia, it may reflect more serious problems. Inadvertently inhaling zinc, which had been a constituent of popular “cold remedies,” has caused anosmia. Another situation where inhaled metal caused anosmia has occurred in welders who routinely inhale fumes containing vaporized iron, chromium, aluminum, and other metals. Head trauma, even from minor injuries, can shear off the olfactory nerves as they pass through the cribriform plate and cause anosmia (see head trauma, Chapter 22).
Another serious problem is that patients with neurodegenerative illnesses lose their sense of smell. For example, almost 90% of patients with Parkinson, dementia with Lewy bodies, Wilson, Creutzfeldt–Jakob, and Alzheimer diseases develop anosmia. In fact, among Parkinson disease patients, more have anosmia than tremor, and it correlates with another manifestation of the illness – dementia. Similarly, anosmia serves as a risk factor for Alzheimer-type dementia. The olfactory bulb manifests the same pathology as the cerebral cortex (see Chapter 7) in Alzheimer and Creutzfeldt–Jakob diseases. Schizophrenic patients also have an increased incidence of anosmia, but not to the degree brought on by neurodegenerative diseases.
Olfactory hallucinations may represent the first phase or aura (Latin, breeze) of complex partial seizures that originate in the medial inferior surface of the temporal lobe. These auras usually consist of several second episodes of ill-defined and unpleasant, but occasionally sweet or otherwise pleasant, smells superimposed on impaired consciousness and behavioral disturbances (see Chapter 10). Also, although most migraine auras consist of visual hallucinations, sometimes olfactory hallucinations represent the aura (see Chapter 9).
Optic (Second)
As for their visual function, the optic nerves originate in visual receptors in the retina and project posteriorly to the optic chiasm. At the chiasm, nasal fibers of the nerves cross, but temporal fibers continue uncrossed (Fig. 4-1). Temporal fibers of one optic nerve join the nasal fibers of the other to form the optic tracts. The tracts pass through the temporal and parietal lobes to terminate in the calcarine cortex of the occipital lobe. Thus, each occipital lobe receives its visual information from the contralateral visual field. Further projections convey the visual information to other areas of the cerebral cortex for decoding work, such as reading, and tracking moving objects.
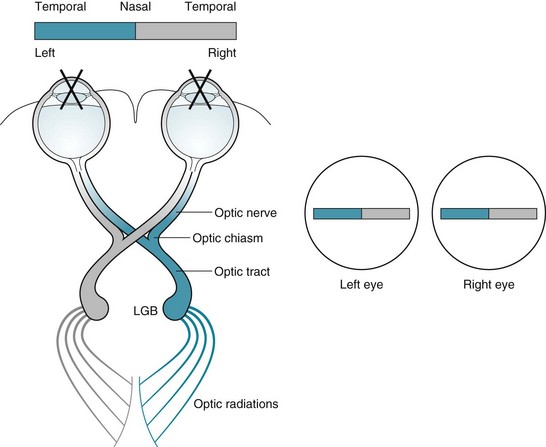
FIGURE 4-1 Left, The optic nerves originate in the retinas. Their medial fibers cross at the optic chiasm while the temporal portions continue uncrossed. The recombinations form the optic tracts that synapse at the lateral geniculate bodies (LGB). The optic tracts sweep through the posterior cerebral hemispheres to terminate at the occipital (“visual”) cortex. This system projects the impulses from each visual field to the contralateral occipital lobe cortex. For example, as in this sketch, impulses conveying the shaded half of the bar, which is in the patient’s left visual field, project to the right occipital cortex. Right, As in this case, medical illustrations typically show a patient’s visual fields from the patient’s perspective. This illustration portrays the shaded portion of the bar in the left visual field of each eye.
Visual field abnormalities are considered among the most important findings in neurology. Many of them point to specific neurologic disorders, such as optic neuritis, pituitary adenomas, and migraines (see Fig. 12-9). In addition, several visual field abnormalities are integral parts of frequently occurring neuropsychiatric conditions, such as left homonymous hemianopsia associated with anosognosia, right homonymous hemianopsia with aphasia or alexia, and incongruous deficits with psychogenic disturbances.
As for their role in regulating the size of the pupil, the optic nerves and tracts form the afferent limb of the pupillary light reflex by sending small branches containing information about light intensity to the midbrain. After a single synapse, the oculomotor nerves (the third cranial nerves) form the efferent limb. The oculomotor nerves, which contain some parasympathetic fibers, innervate the pupils’ constrictor fibers. Overall, the light reflex – optic nerves to midbrain and midbrain to oculomotor nerves – constricts pupil size in response to the intensity of light striking the retina. Simply put, when a neurologist shines a bright light into one or both eyes, the light reflex constricts both pupils (Fig. 4-2).
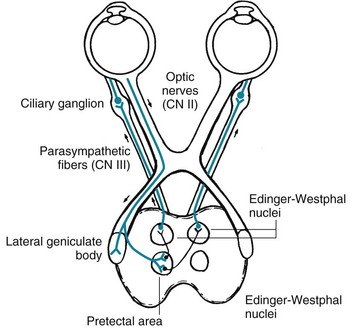
FIGURE 4-2 The light reflex, which is more complex than a deep tendon reflex, begins with its afferent limb in the optic nerve (cranial nerve [CN] II). The optic nerve transmits light impulses from the retinas to two neighboring midbrain structures: (1) In conveying vision, axons synapse on the lateral geniculate body. Then postsynaptic tracts convey visual information to the occipital lobe’s visual cortex. (2) In conveying the light reflex, optic nerve axons also synapse in the pretectal area. Postsynaptic neurons travel a short distance to both the ipsilateral and contralateral Edinger–Westphal nuclei, which are the parasympathetic divisions of the oculomotor (third cranial nerve) nuclei. Those nuclei give rise to parasympathetic oculomotor nerve fibers, which constitute the reflex’s efferent limb (see Fig. 12-17, top). Their fibers synapse in the ciliary ganglia and postsynaptic fibers terminate in the iris constrictor (sphincter) muscles. Thus, light shone in one eye constricts the pupil of that eye (the “direct” [ipsilateral] light reflex) and the contralateral eye (the “consensual” [indirect or contralateral] light reflex). This figure also indicates how oculomotor nerve injuries, because they usually include damage to the parasympathetic component, dilate the pupil. Similarly, it indicates how damaged sympathetic nervous innervation with unopposed parasympathetic innervation, as in the lateral medullary and Horner syndromes, produces pupil constriction (miosis). Finally, it shows how ciliary ganglion damage produces a dilated but extremely sensitive “Adie pupil” (see Fig. 12-17, bottom).
Routine testing of the optic nerve includes examination of: (1) visual acuity (Fig. 12-2); (2) visual fields (Fig. 4-3); and (3) the ocular fundi (Fig. 4-4). Because the visual system is important, complex, and subject to numerous ocular, neurologic, iatrogenic, and psychogenic disturbances, this book dedicates an entire chapter to visual disturbances particularly relevant to psychiatry (see Chapter 12).
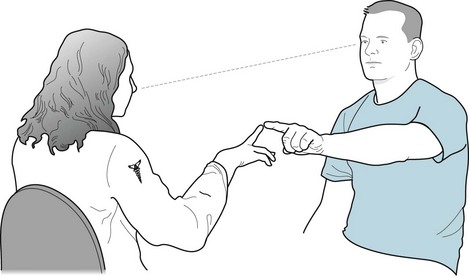
FIGURE 4-3 In testing visual fields by the confrontation method, this neurologist wiggles her index finger as the patient points to it without diverting his eyes from her nose. She tests the four quadrants of each eye’s visual field. (Only by testing each eye individually will she detect a bitemporal quadrantanopia, which is the visual field defect characteristic of pituitary adenomas.) Neurologists test young children and others unable to comply with this method in an abbreviated but still meaningful manner. In this case, the neurologist might assess the response to an attention-catching object introduced to each visual field. For example, a stuffed toy animal, dollar bill, or glass of water should capture a patient’s attention. If the patient does not respond, the neurologist should primarily consider visual field deficit(s). In some cases, a psychogenic visual loss (see later) or inattention (neglect, see Chapter 8) may explain the patient’s failure to respond.
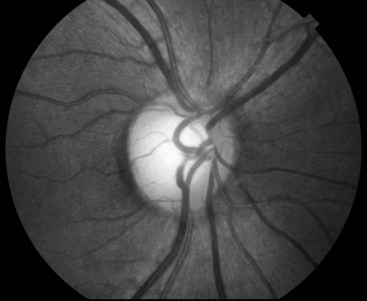
FIGURE 4-4 On fundoscopy, the normal optic fundus or disk appears yellow, flat, and well demarcated from the surrounding red retina. The retinal veins, as everywhere else in the body, are broader than their corresponding arteries. A neurologist will usually see retinal veins pulsate except when intracranial pressure is elevated.
The origin of the optic nerves explains their involvement in certain illnesses and not in others. Unique among the cranial nerves, the optic nerves (and a small proximal portion of the acoustic nerves) are actually projections of the brain coated by myelin derived from oligodendrocytes. In other words, these cranial nerves are extensions of the central nervous system (CNS). Thus, CNS illnesses, particularly childhood-onset metabolic storage diseases and multiple sclerosis (MS)-induced optic neuritis (see Chapter 15), are apt to attack the optic nerves. On the other hand, the optic nerves remain relatively immune from diseases that exclusively attack the peripheral nervous system (PNS) myelin, such as the Guillain–Barré syndrome. Also, by way of contrast, Schwann cells produce the myelin coat of both the remaining cranial nerves and all PNS nerves. They are susceptible to diseases that strike PNS myelin.
Oculomotor, Trochlear, Abducens Nerves (Third, Fourth, Sixth)
The oculomotor nerves (third cranial nerves) originate in the midbrain (Fig. 4-5) and supply the pupil constrictor, eyelid, and adductor and elevator muscles of each eye (medial rectus, inferior oblique, inferior rectus, and superior rectus). Oculomotor nerve impairment, a common condition, thus leads to a distinctive constellation: a dilated pupil, ptosis, and outward deviation (abduction) of the eye (Fig. 4-6). As just discussed, oculomotor nerve injury also impairs the efferent limb of the light reflex. In addition, it impairs the efferent limb of the accommodation reflex, in which the visual system adjusts the shape of the lens to focus on either near or distant objects. (Impaired focusing ability in older individuals, presbyopia [Greek, presbys, old man; opia, eye] results from the aging lens losing its flexibility, not from oculomotor nerve impairment.)
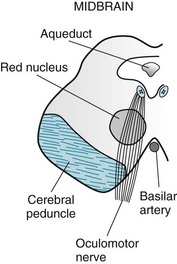
FIGURE 4-5 The oculomotor (third cranial) nerves arise from nuclei in the dorsal portion of the midbrain (see Fig. 2-9). Each descends through the red nucleus, which carries cerebellar outflow fibers to the contralateral limbs. Then each oculomotor nerve passes through the cerebral peduncle, which carries the corticospinal tract destined to innervate the contralateral limbs.
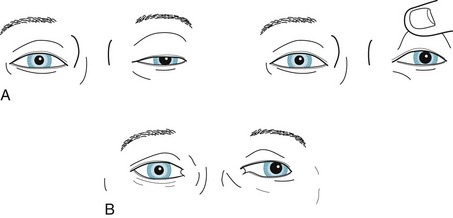
FIGURE 4-6 A, This patient, who is looking ahead, has paresis of the left oculomotor nerve with typical findings: the eye is deviated laterally; the pupil is dilated and unreactive to light; and the upper eyelid covers a portion of the pupil (ptosis). B, In a milder case, with the patient looking ahead, close inspection reveals subtle ptosis, lateral deviation of the eye, and dilation of the pupil. In both cases, patients have diplopia that increases when looking to the right because this movement requires adducting the left eye, but the paretic left medial rectus muscle cannot participate and the patient’s gaze becomes dysconjugate (see Fig. 12-13).
Unlike the third and fourth cranial nerves, the abducens nerves (sixth cranial nerves) originate in the pons (Fig. 4-7 and see Fig. 2-9). Like the fourth cranial nerves, the abducens nerves perform only a single function and innervate only a single muscle. Each abducens nerve innervates its ipsilateral lateral rectus muscle, which abducts the eyes. Abducens nerve impairment, which is relatively common, leads to inward deviation (adduction) of the eye from the unopposed medial pull of the oculomotor nerve, but no ptosis or pupil changes (Fig. 4-8). To review: the lateral rectus muscle is innervated by the sixth cranial (abducens) nerve and the superior oblique by the fourth (trochlear), but all the others by the third (oculomotor). A mnemonic device, “LR6SO4,” captures this relationship.
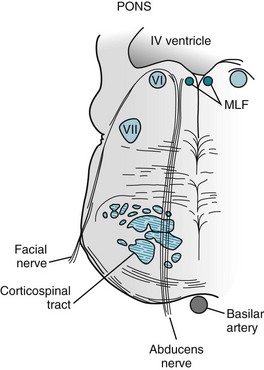
FIGURE 4-7 The abducens (sixth cranial) nerves arise from nuclei located in the dorsal portion of the pons. These nuclei are adjacent to the medial longitudinal fasciculus (MLF; see Fig. 15-3). As the abducens nerves descend, they pass medial to the facial nerves, and then between the upper motor neurons of the corticospinal tract.

FIGURE 4-8 This patient with paresis of the left abducens nerve has medial deviation of the left eye. He will have diplopia on looking ahead and toward the left, but not when looking to the right (see Fig. 12-14).
For learning purposes, neurologists might best consider ocular cranial nerve lesions according to their brainstem level (midbrain and pons) and correlate clinical features with the admittedly complex anatomy. Because the anatomy is so compact, brainstem lesions that damage cranial nerves typically produce classic combinations of injuries of the ocular nerves and the adjacent corticospinal (pyramidal) tract or cerebellar outflow tracts. These lesions cause diplopia accompanied by contralateral hemiparesis or ataxia. The pattern of the diplopia is the signature of the lesion’s location. The etiology in almost all cases is an occlusion of a small branch of the basilar artery causing a small brainstem infarction (see Chapter 11).
Most importantly, despite producing complex neurologic deficits, brainstem lesions generally do not impair cognitive function. Nevertheless, certain exceptions to this dictum bear mentioning. Wernicke encephalopathy, for example, consists of memory impairment (amnesia) accompanied by nystagmus and oculomotor or abducens nerve impairment (see Chapter 7). Another exception is transtentorial herniation, in which a cerebral mass lesion, such as a subdural hematoma, squeezes the anterior tip of the temporal lobe through the tentorial notch. In this situation, the mass compresses the oculomotor nerve and brainstem to cause coma, decerebrate posturing, and a dilated pupil (see Fig. 19-3).
The following frequently occurring, classic brainstem syndromes, despite their pronounced deficits, typically spare cognitive function. With a right-sided midbrain infarction a patient would have a right oculomotor nerve palsy, which would cause right ptosis, a dilated pupil, and diplopia, accompanied by left hemiparesis (Fig. 4-9). With a slightly different right-sided midbrain infarction, a patient might have right oculomotor nerve palsy and left tremor (Fig. 4-10).
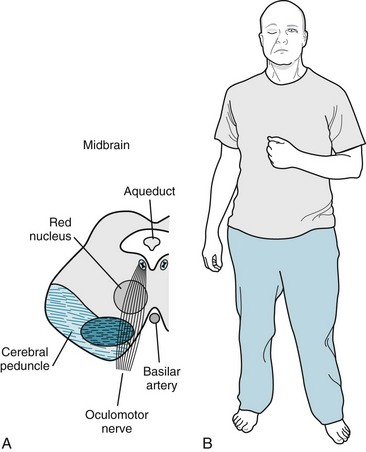
FIGURE 4-9 A, A right midbrain infarction damages the oculomotor nerve that supplies the ipsilateral eye and the adjacent cerebral peduncle, which contains the corticospinal tract that subsequently crosses in the medulla and ultimately supplies the contralateral arm and leg. B, This patient has right-sided ptosis from the right oculomotor nerve palsy and left hemiparesis from the corticospinal tract injury. Also note that the ptosis elicits a compensatory unconscious elevation of the eyebrow to uncover the eye. Neurologists also see eyebrow elevation in other conditions that cause ptosis, such as the lateral medullary syndrome (Fig. 2-10), myasthenia gravis (Fig. 6-3), and cluster headache (Fig. 9-4).
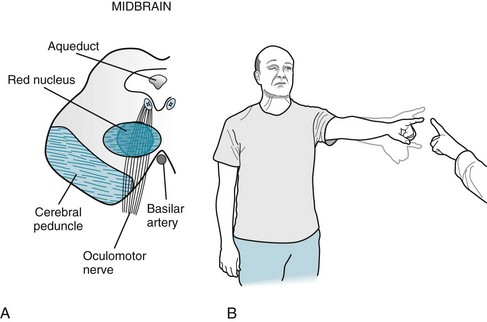
FIGURE 4-10 A, The red nucleus is the intermediate step in conveying cerebellar outflow from the cerebellum to the ipsilateral arm and leg. Each cerebellar hemisphere innervates the contralateral red nucleus that, in turn, innervates the contralateral arm and leg. Because this pattern involves two contralateral steps, neurologists often call it the “double cross.” In this case, a right midbrain infarction damages the oculomotor nerve and adjacent red nucleus, which innervates the left arm and leg. B, This patient has right ptosis from the oculomotor nerve palsy and left arm ataxia from the damage to the cerebellar outflow tract.
A right-sided pons lesion typically translates into a right abducens nerve paresis and left hemiparesis (Fig. 4-11). Notably, in each of these brainstem injuries, mental status remains normal because the cerebrum is unscathed.
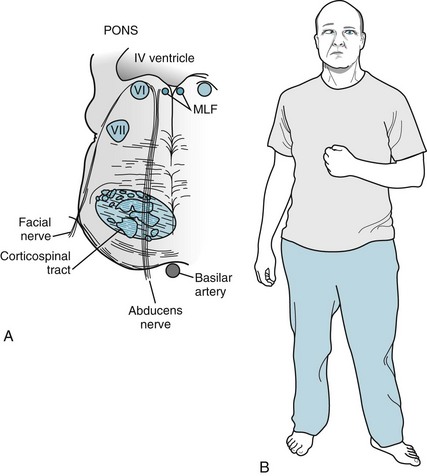
FIGURE 4-11 A, A right pontine infarction damages the abducens nerve, which supplies the ipsilateral eye, and the adjacent corticospinal tract, which supplies the contralateral limbs. (This situation is analogous to midbrain infarctions: Fig. 4-8.) B, This patient has inward deviation of the right eye from paresis of the right abducens nerve, and left hemiparesis from right corticospinal tract damage. MLF, medial longitudinal fasciculus.
Another common site of brainstem injury that affects ocular motility is the medial longitudinal fasciculus (MLF). This structure is the heavily myelinated midline tract between the pons and the midbrain that links the nuclei of the abducens and oculomotor nerves (see Figs 2-9, 4-11, 15-3, and 15-4). Its interruption produces the MLF syndrome, also called internuclear ophthalmoplegia, which consists of nystagmus of the abducting eye and failure of the adducting eye to cross the midline. This disorder is best known as a characteristic sign of MS.
Ruptured or expanding aneurysms of the posterior communicating artery may compress the oculomotor nerve, just as it exits from the midbrain. In this case, oculomotor nerve palsy – which would be the least of the patient’s problems – is just one manifestation of a life-threatening subarachnoid hemorrhage that usually renders patients prostrate from a headache. Children occasionally have migraine headaches accompanied by temporary oculomotor nerve paresis (see Chapter 9). By way of contrast, in motor neuron diseases, amyotrophic lateral sclerosis (ALS) and poliomyelitis, the oculomotor and abducens nerves retain normal function despite destruction of large numbers of motor neurons. Patients may have full, conjugate eye movements despite being unable to breathe, lift their limbs, or move their head.
Disorders of the neuromuscular junction – the cranial and peripheral nerve’s furthest extent – also produce oculomotor or abducens nerve paresis. In myasthenia gravis (see Fig. 6-3) and botulism, for example, impaired acetylcholine neuromuscular transmission leads to combinations of ocular and other cranial nerve paresis. These deficits may puzzle neurologists because the muscle weakness is often subtle and variable in severity and pattern. Neurologists may overlook mild cases or misdiagnose them as a psychogenic disorder. Nevertheless, they illustrate neuroanatomic relationships and are clinically important, especially in their extremes. For example, severe cases may lead to respiratory impairment.
Trigeminal (Fifth)
Examination of the trigeminal nerve begins by testing sensation in its three sensory divisions (Fig. 4-12). Neurologists touch the side of the patient’s forehead, cheek, and jaw. Areas of reduced sensation, hypalgesia, should conform to anatomic outlines.
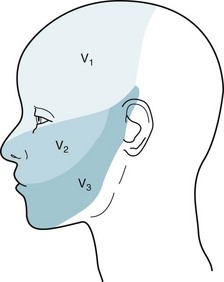
FIGURE 4-12 The three divisions of the trigeminal nerve convey sensory innervation of the face. The first division (V1) supplies the forehead, the cornea, and the scalp up to the vertex; the second (V2) supplies the malar area; and the third (V3) supplies the lower jaw, except for the angle. These dermatomes hold more than academic interest. Herpes zoster infections (shingles), trigeminal neuralgia (see Chapter 9), and facial angioma in the Sturge–Weber syndrome (see Fig. 13-13) each typically affect one or another dermatome. In contrast, psychogenic disturbances do not confine themselves to a single dermatome.
In testing the trigeminal nerve’s motor component, neurologists assess jaw muscle strength by asking the patient to clench and then protrude the jaw. The jaw jerk reflex consists of a prompt but not overly forceful closing after a tap (Fig. 4-13). Alterations in the response follow the rules of a deep tendon reflex (DTR). A hyperactive response indicates an UMN (corticobulbar tract) lesion, and a hypoactive response indicates a lower motor neuron (LMN) or cranial nerve lesion. The neurologist should include testing of the jaw jerk reflex in patients with dysarthria, dysphagia, and emotional lability – mostly to assess them for the likelihood of pseudobulbar palsy (see later).
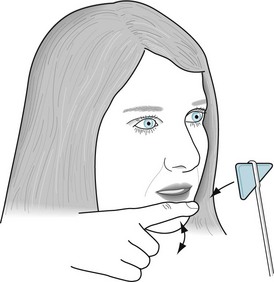
FIGURE 4-13 Tapping the normal, open, relaxed jaw will move it slightly downward. The jaw jerk reflex is the soft rebound. Abnormalities are mostly a matter of the rebound’s rapidity and strength. In a hypoactive reflex, as found in bulbar palsy and other lower motor neuron injuries, patients show little or no rebound. In a hyperactive reflex, as in pseudobulbar palsy and other upper motor neuron (corticobulbar tract) lesions, patients show a quick and forceful rebound.
Injury of a trigeminal nerve causes facial hypalgesia, afferent corneal reflex impairment, jaw jerk hypoactivity, and deviation of the jaw toward the side of the lesion. A variety of conditions – nasopharyngeal tumors, gunshot wounds, and tumors of the cerebellopontine angle, such as acoustic neuromas (see Fig. 20-27) – may cause trigeminal nerve injury.
In a frequently occurring situation, an aberrant vessel or other lesion in the cerebellopontine angle, MS plaques in the pons, or unknown disorder irritates the trigeminal nerve. The irritation causes a terribly painful condition, trigeminal neuralgia, in which patients suffer lancinating jabs in the distribution of a single division of one nerve (see Chapter 9). Similarly, when herpes zoster infects the trigeminal nerve it causes a rash followed by excruciating pain (postherpetic neuralgia) in the distribution of a single division of one trigeminal nerve (see Chapter 14).
Finally, a psychogenic sensory loss involving the face will usually encompass the entire face or be included in a sensory loss of one-half of the body, i.e., a hemisensory loss. In almost all cases, the following three nonanatomic features will be present: (1) the sensory loss will not involve the scalp (although the portion anterior to the vertex is supplied by the trigeminal nerve); (2) the corneal reflex will remain intact; and (3) when only one-half of the face is affected, sensation will be lost sharply rather than gradually at the midline (i.e., the patient will “split the midline”) (see Fig. 3-5).
Facial (Seventh)
Just as the trigeminal nerves supply the muscles of mastication, the facial nerves supply the “muscles of facial expression.” In a unique and potentially confusing arrangement in their neuroanatomy, cerebral impulses innervate both the contralateral and ipsilateral facial nerve motor nuclei. Each facial nerve supplies its ipsilateral temporalis, orbicularis oculi, and orbicularis oris muscles – muscles responsible for a frown, raised eyebrows, wink, smile, and grimace. In the classic explanation, because of their crossed and uncrossed supply, the upper facial muscles are essentially innervated by both cerebral hemispheres, whereas the lower facial muscles are innervated by only the contralateral cerebral hemisphere (Fig. 4-14). A newer explanation proposes that interneurons in the brainstem link the facial nerve nuclei.
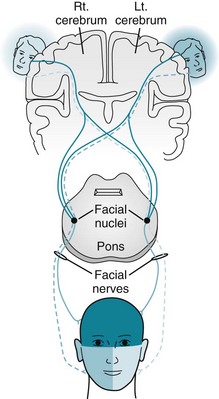
FIGURE 4-14 In the classic portrayal, corticobulbar tracts originating in the ipsilateral, as well as in the contralateral, cerebral hemisphere supply each facial nerve nucleus. Each facial nerve supplies the ipsilateral muscles of facial expression. Because the upper half of the face receives cortical innervation from both hemispheres, cerebral injuries lead to paresis only of the lower half of the contralateral face. In contrast, facial nerve injuries lead to paresis of both the upper and lower halves of the ipsilateral side of the face.
Facial nerve damage typically produces paresis of the ipsilateral upper and lower face muscles with or without loss of taste sensation. Sudden onset, idiopathic facial paralysis, usually with loss of taste sensation, generically labeled Bell’s palsy, has traditionally been attributed to an inflammation or infection of the nerve (Fig. 4-15). In many of these cases, herpes simplex virus or, less often, Borrelia burgdorferi, a tick-borne spirochete that causes Lyme disease (see Chapters 5 and 7), has been the culprit. Destructive injuries, including lacerations, cerebellopontine angle tumors, and carcinomatous meningitis, damage not only the facial nerve, but usually also its neighboring cerebellopontine angle nerves.
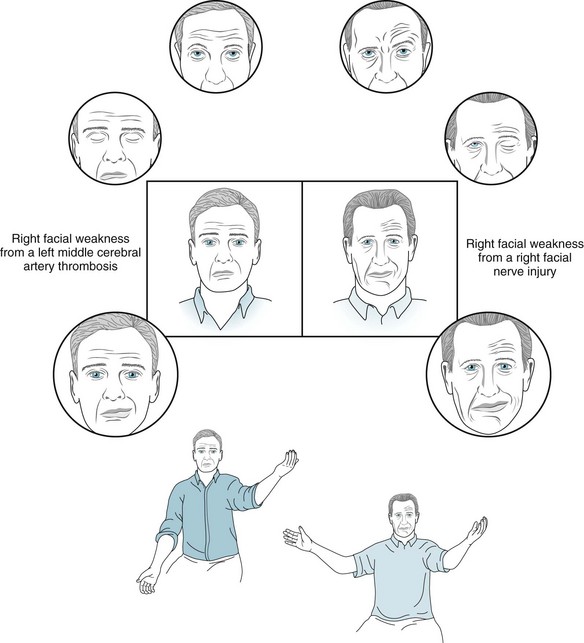
FIGURE 4-15 The man on the left has weakness of his right lower face from thrombosis of the left middle cerebral artery: Neurologists might say that he has a “central” (central nervous system: CNS) facial paralysis. In contrast, the man on the right has right-sided weakness of both his upper and lower face from a right facial nerve injury (Bell’s palsy): Neurologists might say that he has a “peripheral” facial (cranial nerve) paralysis. In the center boxed sketches, the man with the central palsy (left) has flattening of the right nasolabial fold and sagging of the mouth downward to the right. This pattern of weakness indicates paresis of only the lower facial muscles. The man with the peripheral palsy (right), however, has right-sided loss of the normal forehead furrows in addition to flattening of his nasolabial fold. This pattern of weakness indicates paresis of the upper as well as the lower facial muscles. The neurologist has asked the men in the circled sketches at the top to look upward – a maneuver that would exaggerate upper facial weakness. The man with central weakness has normal upward movement of the eyebrows and furrowing of the forehead. The man with peripheral weakness has no eyebrow or forehead movement, and the forehead skin remains flat. The neurologist has asked the men in the circled sketches second from the top to close their eyes – a maneuver that also would exaggerate upper facial weakness. The man with the central weakness has widening of the palpebral fissure, but he is able to close his eyelids and cover the eyeball. The man with the peripheral weakness is unable to close the affected eyelid, although his genuine effort is made apparent by the retroversion of the eyeball (Bell’s phenomenon). The neurologist has asked the men in the lowest circled sketches to smile – a maneuver that would exaggerate lower facial weakness. Both men have strength only of the left side of the mouth, and thus it deviates to the left. If tested, the man with Bell’s palsy would have loss of taste on the anterior two-thirds of his tongue on the affected side. The neurologist in the bottom sketches has asked both men to elevate their arms. The man with the central facial weakness also has paresis of the adjacent arm, but the man with the peripheral weakness has no arm paresis. In summary, the man on the left with the left middle cerebral artery occlusion has paresis of his right lower face and arm. The man on the right with right Bell’s palsy has paresis of his right upper and lower face and loss of taste on the anterior two-thirds of his tongue.
Lesions that stimulate the nerve have the opposite effect. For example, aberrant vessels in the cerebellopontine angle can irritate the facial nerve and produce intermittent, completely involuntary, prolonged contractions of the muscles of the ipsilateral side of the face. This disorder, hemifacial spasm (see Chapter 18), which casual observers might misdiagnose as a “nervous tic,” represents the facial nerve counterpart of trigeminal neuralgia.
Acoustic (Eighth)
Each acoustic nerve is composed of two divisions with separate courses and functions: hearing and balance. The cochlear nerve, one of the two divisions, transmits auditory impulses from each middle and inner ear mechanism to the superior temporal gyri of both cerebral hemispheres (Fig. 4-16). This bilateral cortical representation of sound explains the everyday observation that damage to the ear or acoustic nerve may cause deafness in that ear, but the patient will still hear sound and speech because it passes through the other acoustic nerve or ear. The bilateral cortical representation of sound also explains why unilateral lesions of the brainstem or cerebral hemisphere – CNS damage – will not cause it. For example, cerebral lesions, such as tumors or strokes, that involve the temporal lobes may cause aphasia and hemiparesis, but they do not impair hearing.
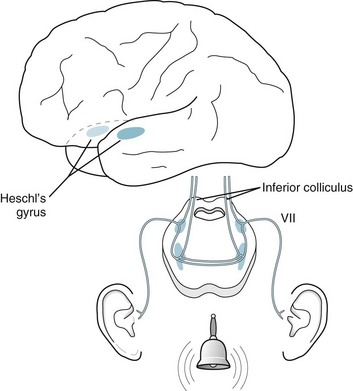
FIGURE 4-16 The cochlear division of the acoustic nerve synapses extensively in the pons. Crossed and uncrossed fibers pass upward through the brainstem to terminate in the ipsilateral and contralateral auditory (Heschl gyrus) cortex of each temporal lobe; however, Heschl gyri, which sit in the planum temporale, receive auditory stimuli predominantly from the contralateral ear. In addition, the dominant-hemisphere Heschl gyrus almost abuts Wernicke language area (see Fig. 8-1) and predictably has a major role in language function.
Acoustic nerve injury may result from medications, such as aspirin or streptomycin, skull fractures severing the nerve, or cerebellopontine angle tumors, particularly acoustic neuromas associated with neurofibromatosis (see Chapter 13). Although cognitive impairment does not generally accompany deafness, in utero rubella infections or kernicterus (see Chapter 13) commonly cause syndromes of mental retardation and congenital deafness. In a related situation, children with congenital hearing impairment, deprived of proper intervention, may grow up to appear mentally retarded and have some features of autism. Cochlear implants, a unique, life-improving innovation, have allowed hearing-impaired infants and children to develop hearing and speaking abilities, such that most of them can enter mainstream education.
As with many age-related impairments, presbycusis results more from degeneration of the special sensory organ than the cranial nerve itself (Box 4-2). In this case, the cochlear mechanism, rather than the acoustic nerve itself, withers. Presbycusis potentially leads to inattention and social isolation. In addition, when hearing impairment accompanies visual impairments, the resulting sensory deprivation may precipitate hallucinations. Such impairments may also overwhelm someone with mild cognitive impairment (see Chapter 7) and lead to misdiagnoses of dementia and psychosis. For the limited problem of age-related hearing impairment in the elderly, physicians should as a general rule dispense hearing aids readily and even on a trial basis. In some cases, cochlear implants have allowed adults with hearing loss uncorrected by hearing aids to regain useful hearing; however, unlike infants and young children, adults usually cannot learn to translate the electric impulses into comprehensible speech.
Box 4-2
Age-Related Special Sense Impairments
| Smell | Some degree of anosmia in 75% of individuals older than 80 years |
| Vision | Presbyopia: mostly inability to accommodate to see closely held or small objects; cataracts (see Chapter 12) |
| Taste | Loss of taste sensitivity and discrimination, as well as anosmia for aroma |
| Hearing | Presbyacusis: loss of speech discrimination, especially for consonants; poor high-pitched sound detection, tinnitus |
When patients seem to mimic deafness, neurologists may attempt to startle them with a loud sound or watch for an auditory-ocular reflex (involuntarily looking toward a noise). Neurologists wishing to confirm a diagnosis of psychogenic hearing loss may order brainstem auditory-evoked response (BAER) testing (see Chapter 15). Audiologic testing is advisable in children with autism, cerebral palsy, mental retardation, speech impediments, and poor school performance, as well as those suspected of having a psychogenic hearing impairment.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


