Chapter 7 Dementia
Neurologists and most other physicians continue to use the term dementia, which they see as a clinical condition or syndrome of a progressive decline in cognitive function that impairs daily activities. Neurologists require memory impairment plus one or more of the following: aphasia, apraxia, agnosia, or disturbance in executive function (see Chapter 8). Because their definition requires two domains, it excludes isolated amnesia (Greek, forgetfulness) or aphasia (Greek, speechlessness).
Disorders Related to Dementia
Congenital Cognitive Impairment
Physical manifestations of congenital cerebral injury, such as seizures and hemiparesis (“cerebral palsy,” see Chapter 13), frequently accompany Intellectual Developmental Disorder. In cases of genetic abnormalities, distinctive behavioral disturbances and anomalies of nonneurologic organs – the face, skin, ocular lenses, kidneys, and skeleton – often form syndromes.
Amnesia
Neurologists usually attribute amnesia to transient or permanent dysfunction of the hippocampus (Greek, sea horse) and other portions of the limbic system, which are based in the temporal and frontal lobes (see Fig. 16-5). Dementia, in contrast, usually results from extensive cerebral cortex dysfunction (see later).
Transient amnesia is an important, relatively common disturbance that usually consists of a suddenly occurring period of amnesia lasting only several minutes to several hours. It has several potential medical and neurologic explanations (Box 7-1). One of them, electroconvulsive therapy (ECT), routinely induces both anterograde and retrograde amnesia, with retrograde amnesia, especially for autobiographic information, tending to persist longer than anterograde amnesia. ECT-induced amnesia is more likely to occur or to be more pronounced following treatment with high electrical dosage, with bilateral rather than unilateral electrode placement, with use of alternating current, and three-times rather than two-times weekly administration. Without a pretreatment assessment of a patient’s memory and other aspects of cognitive function, clinicians may have problems separating ECT-induced amnesia from memory difficulties reflecting underlying depression, medications, and, especially in the elderly, pre-existing cognitive impairment.
Box 7-1
Commonly Cited Causes of Transient Amnesia
Various neuropsychologic and physical abnormalities usually accompany amnesia from neurologic conditions. For example, behavioral disturbances, depression, and headache are comorbid with posttraumatic amnesia. With severe traumatic brain injury (TBI, see Chapter 22), hemiparesis, ataxia, pseudobulbar palsy, or epilepsy accompanies posttraumatic amnesia. Similarly, in addition to its characteristic anterograde amnesia, the Wernicke–Korsakoff syndrome comprises ataxia and signs of a peripheral neuropathy (see later).
In another example, herpes simplex encephalitis causes amnesia accompanied by personality changes, complex partial seizures, and the Klüver–Bucy syndrome (see Chapters 12 and 16) because the virus typically enters the undersurface of the brain through the nasopharynx and attacks the frontal and temporal lobes. This condition occurs relatively frequently because herpes simplex is the most common cause of sporadically occurring (nonepidemic) viral encephalitis. (Human immunodeficiency virus [HIV] encephalitis, which does not cause this scenario, is epidemic.)
Conversely, apparent memory impairment may also appear as an aspect of several psychiatric disorders (see Chapter 11, dissociative amnesia). In general, individuals with amnesia from psychiatric illness or malingering (nonneurologic amnesia) lose memory for personal identity or emotionally laden events rather than recently acquired information. For example, a criminal deeply in debt may travel to another city and “forget” his debts, wife, and past associates, but he would retain his ability to recall people, events, and day-to-day transactions in his new life. Nonneurologic amnesia also characteristically produces inconsistent results on formal memory testing. In a simple example, a workman giving emphasis to the sequelae of a head injury may seem unable to recall three playing cards after 30 seconds, but half an hour after discussing neutral topics will recall three different cards after a 5-minute interval. Also, Amytal infusions may temporarily restore memories in individuals with nonneurologic amnesia, but not in those with brain damage.
Neuropsychologic Conditions
Confabulation is a neuropsychologic condition in which patients offer implausible explanations in a sincere, forthcoming, and typically jovial manner. Although individuals with confabulation disregard the truth, they do not intentionally deceive. Confabulation is a well-known aspect of Wernicke–Korsakoff syndrome, Anton syndrome (see cortical blindness, Chapter 12), and anosognosia (see Chapter 8). With these conditions referable to entirely different regions of the brain, confabulation lacks consistent anatomic correlations and physical features.
Discrete neuropsychologic disorders – aphasia, anosognosia, and apraxia – may occur alone, in various combinations, or as comorbidities of dementia (see Chapter 8). If one of them occurs with a comorbidity of dementia, it indicates that the dementia originates in “cortical” rather than “subcortical” dysfunction (see later). These disorders, unlike dementia, are attributable to discrete cerebral lesions. Sometimes only neuropsychologic testing can detect these disorders and tease them apart from dementia.
Normal Aging
Sleep
Among the elderly, times of falling asleep and awakening both phase-advance (occur earlier than usual), slow-wave sleep declines, and sleep fragments. In addition, restless legs syndrome and rapid eye movement (REM) behavioral disorder commonly disrupt their sleep (see Chapter 17). All these changes may occur whether or not the elderly have dementia.
Special Senses
Age-related deterioration of sensory organs impairs hearing and vision (see Chapter 12). Older individuals typically have small, less reactive pupils and some retinal degeneration. They require greater light, more contrast, and sharper focusing to be able to read and drive. Their hearing tends to be poorer, especially for speech discrimination. Their senses of taste and smell also deteriorate.
EEG and Imaging Changes
As individuals age, the electroencephalogram (EEG) background alpha activity slows to the lower end of the normal 8–12-Hz alpha range. Computed tomography (CT) and magnetic resonance imaging (MRI) often reveal decreased volume of the frontal and parietal lobes, atrophy of the cerebral cortex, expansion of the Sylvian fissure, and concomitantly increased volume of the lateral and third ventricles (see Figs 20-2, 20-3, and 20-18). In addition, MRI reveals white-matter hyperintensities (“white dots”). Although striking, these abnormalities are usually innocuous and, by themselves, do not reflect the onset of Alzheimer disease.
Dementia
Classifications and Causes
• Prevalence: Studies reporting on the epidemiology of dementia-producing illnesses vary by whether cases were clinical or postmortem, drawn from primary or tertiary care settings, or if the criteria were incidence or prevalence. By any measure, Alzheimer disease is the most prevalent cause of dementia, not only because its incidence is so high, but also, with its victims living for a relatively long time, its prevalence is also high. A typical breakdown of the prevalence of dementia-producing illnesses would list Alzheimer disease 70%, dementia with Lewy bodies (DLB) 15%, vascular cognitive impairment (VCI) or the preliminary DSM-5 term Vascular Neurocognitive Impairment 10%, frontotemporal dementia 5–10%, and all others, in total, 5–10%.
• Patient’s age at the onset of dementia: Similarly, beginning at age 65 years, those same illnesses cause almost all cases of dementia. However, individuals between 21 and 65 years are liable to succumb to different dementia-producing illnesses: HIV disease, substance abuse, severe TBI, end-stage multiple sclerosis (MS) (see Chapter 15), frontotemporal dementia, and VCI. In adolescence, the causes are different and more numerous (Box 7-2).
• Accompanying physical manifestations: Distinctive physical neurologic abnormalities that may accompany dementia and allow for a diagnosis by inspection include ocular motility impairments, gait apraxia (see later), myoclonus (see later and Chapter 18), peripheral neuropathy (see Box 5-1), chorea, other involuntary movement disorders (see Box 18-4), and lateralized signs, such as hemiparesis.
• Genetics: Of frequently occurring illnesses, Huntington disease, frontotemporal dementia, some prion illnesses, and, in certain families, Alzheimer disease follow an autosomal dominant pattern. Wilson disease follows an autosomal recessive pattern.
• Rapidity of onset: In patients with Alzheimer disease, dementia evolves over a period of many years to a decade, which serves as the reference point. In contrast, several diseases produce dementia within 6–12 months. These “rapidly progressive dementias” include HIV-associated dementia, frontotemporal dementia, dementia with Lewy bodies (see later), paraneoplastic limbic encephalitis (see Chapter 19), and, perhaps most notoriously, Creutzfeldt–Jakob disease and its variant.
• Reversibility: The most common conditions that neurologists usually list as “reversible causes of dementia” are depression, over-medication, hypothyroidism, B12 deficiency, other metabolic abnormalities, subdural hematomas, and normal-pressure hydrocephalus (NPH). Although reversible dementias are rightfully sought, the results of treatment are discouraging. Only about 9% of dementia cases are potentially reversible and physicians actually partially or fully reverse less than 1%.
• Cortical and subcortical dementias: According to a cortical/subcortical distinction, cortical dementias consist of illnesses in which neuropsychologic signs of cortical injury – typically aphasia, agnosia, and apraxia – accompany dementia. Because the brain’s subcortical areas are relatively untouched, patients remain alert, attentive, and ambulatory. Alzheimer disease serves as the prime example of a cortical dementia.
Box 7-2
Causes of Dementia in Adolescents
Mental Status Testing
Mini-Mental State Examination (MMSE) (Fig. 7-1)

FIGURE 7-1 Mini-Mental State Examination (MMSE). Points are assigned for correct answers. Scores of 20 points or less indicate dementia, delirium, schizophrenia, or affective disorders – alone or in combination. However, such low scores are not found in normal elderly people or in those with neuroses or personality disorders. MMSE scores fall in proportion to dementia-induced impairments in daily living as well as cognitive decline. For example, scores of 20 correlate with inability to keep appointments or use a telephone. Scores of 15 correlate with inability to dress or groom.
(Adapted from Folstein MF, Folstein SE, McHugh PR. “Mini-Mental state:” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. © 1975, 1998 MiniMental LLC.)
Alzheimer Disease Assessment Scale (ADAS)
The ADAS consists of cognitive and noncognitive sections. Its cognitive section (ADAS-Cog) (Fig. 7-2) includes not only standard tests of language, comprehension, memory, and orientation, but also tests of visuospatial ability, such as drawing geometric figures, and physical tasks that reflect ideational praxis, such as folding a paper into an envelope. Patients obtain scores of 0–70 points in proportion to worsening performance.
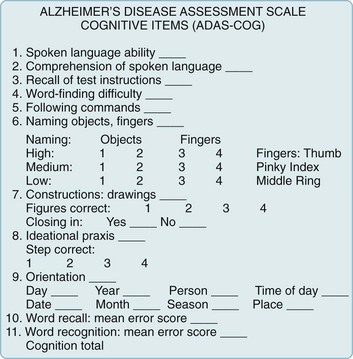
FIGURE 7-2 The cognitive section of the Alzheimer Disease Assessment Scale (ADAS-Cog), the standard test for assessing pharmacologic intervention, has been designed to measure many important aspects of cognitive function that are apt to deteriorate in Alzheimer disease. The noncognitive section measures mood, attention, delusions, and motor activity, such as pacing and tremors. As dementia begins or worsens, patients’ responses change from no impairment (where the patient receives 0 points) to severe cognitive impairment (5 points). In other words, as dementia worsens, patients accumulate points. Total ADAS-Cog scores for patients with mild to moderate Alzheimer disease range from 15 to 25 and, as cognitive function declines, scores increase by 6–12 points yearly.
(From Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry 1984;141:1356–1364. Reprinted with permission from the American Journal of Psychiatry, Copyright 1984. American Psychiatric Association.)
Montreal Cognitive Assessment (MoCA) (Fig. 7-3)
The MoCA, which has greater sensitivity than the MMSE, readily detects mild impairment. It is useful when a patient’s only symptom is memory impairment or physicians suspect cognitive impairment in individuals who score 25 points or better on the MMSE. Neurologists also administer the MoCA to patients with any neurodegenerative illness – a category that includes Alzheimer, Parkinson, and dementia with Lewy bodies diseases, and frontotemporal dementia. In Parkinson disease, the MoCA compared to the MMSE is more sensitive in detecting early cognitive impairment (see Chapter 18).

FIGURE 7-3 The Montreal Cognitive Assessment (MoCA). This assessment includes eight cognitive domains. As with the MMSE, the total score reaches 30 and values lower than 26 indicate cognitive impairment. However, some authors found that a cut-off of 23 gave a sensitivity and specificity of at least 95%.
(Copyright Z. Nasreddine MD. Reproduced with permission. The test and instruction may be accessed at www.mocast.org.)
Further Testing
• For very intelligent, well-educated, or highly functional individuals with symptoms compatible with dementia whose screening tests fail to show a cognitive impairment.
• For individuals whose ability to execute critical occupational or personal decisions, including legal competency, must be assured.
• For assessing individuals with confounding deficits, such as intellectual development disorder, learning disabilities, minimal education, deafness, or aphasia.
• In distinguishing dementia from depression, other psychiatric disturbances, and malingering.
• In distinguishing Alzheimer disease from frontotemporal dementia.
Laboratory Evaluation in Dementia
Depending on the clinical evaluation, neurologists generally request a series of laboratory tests (Box 7-3). Although testing is expensive (see Appendix 2), it may allow a firm diagnosis or even detect a potentially correctable cause of dementia. In addition, certain tests may reveal the illness before individuals manifest cognitive impairment, i.e., in their preclinical or presymptomatic state. Neurologists modify the testing protocol if dementia has developed in an adolescent (see Box 7-2) or has progressed rapidly.
Physicians should reserve certain other tests for particular indications. For instance, if adolescents or young adults develop dementia, an evaluation might include serum ceruloplasmin determination and slit-lamp examination for Wilson disease; urine toxicology screens for drug abuse; and, depending on the circumstances, urine analysis for metachromatic granules and arylsulfatase-A activity for metachromatic leukodystrophy (see Chapter 5). Likewise, physicians should judiciously request serologic tests for autoimmune or inflammatory disease, serum Lyme disease titer determinations, and other tests for systemic illnesses.
Alzheimer Disease
Dementia
In the dementia stage, patients may remain superficially conversant, sociable, able to perform routine tasks, and physically intact. Nevertheless, the spouse or caregiver will report that they suffer from incapacitating memory impairment and inability to cope with new situations. Patients’ language impairment typically includes a decrease in spontaneous verbal output, an inability to find words (anomia), use of incorrect words (paraphasic errors), and a tendency to circumvent forgotten words – elements of aphasia (see Chapter 8).
Neuropsychiatric Manifestations
Wandering constitutes a particularly troublesome, although not unique, manifestation of Alzheimer disease. It may originate in any combination of numerous disturbances, including memory impairment, visuospatial perceptual difficulties, delusions and hallucinations, akathisia (see Chapter 18), and mundane activities, such as looking for food.
Alzheimer patients also lose their normal circadian sleep–wake pattern to a greater degree than cognitively intact elderly people (see Chapter 17). Their sleep becomes fragmented throughout the day and night. The disruption of their sleep parallels the severity of their dementia.
Physical Signs
Patients with Alzheimer disease characteristically have no physical impairment, except for one small but intriguing finding – anosmia (see Chapter 4). Even before the onset of dementia, Alzheimer disease patients require high concentrations of a volatile substance to detect and identify it. Neurologists also find anosmia in patients with Parkinson disease and dementia with Lewy bodies disease, but not in those with VCI or depression. Patients with TBI with or without dementia also have anosmia because they have had shearing of the olfactory nerve fibers at the cribriform plate (see Chapter 22).
Until Alzheimer disease advances to unequivocal dementia, patients usually remain ambulatory and able to feed themselves. The common sight of an Alzheimer disease patient walking steadily but aimlessly through a neighborhood characterizes the disparity between intellectual and motor deficits. When patients reach advanced stages of the illness, physicians can elicit frontal release signs (Fig. 7-4), increased jaw jerk reflex (see Fig. 4-13), and Babinski signs. Unlike patients with VCI, those with Alzheimer disease do not have lateralized signs. They eventually become mute, fail to respond to verbal requests, remain confined to bed, and assume a decorticate (fetal) posture. They frequently slip into a persistent vegetative state (see Chapter 11).
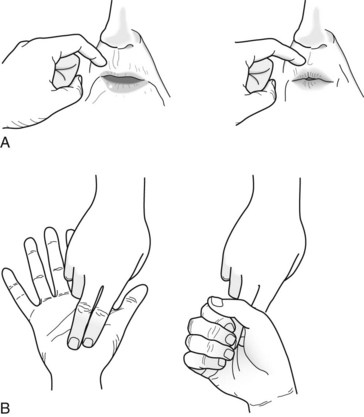
FIGURE 7-4 The snout and grasp reflexes – frontal lobe release reflexes – are frequently present in individuals with severe dementia. A, Physicians elicit the snout reflex by tapping the patient’s upper lip with a finger or a percussion hammer. This reflex causes the patient’s lips to purse and lower facial muscles to pout. B, Physicians elicit the grasp reflex by stroking the patient’s palm crosswise or the fingers lengthwise. The reflex causes the patient to grasp the physicians’ fingers and fail to release despite requests.
Laboratory Tests
In about one-half of Alzheimer patients, PET for glucose metabolism shows areas of decreased metabolism in the bilateral parietal and temporal association cortex (see Fig. 20-29). These hypometabolic areas are vague at first, but as the disease progresses, hypometabolism spreads to the frontal lobe cortex and becomes more distinct. PET remains unsuitable for routine testing, mostly because of its low sensitivity and specificity, as well as its cost. The changes are more specific and more cost-effective for frontotemporal dementia (see later). PET for amyloid accumulation is useful in the diagnosis of the preclinical stage of Alzheimer disease, but it is probably unnecessary in the symptomatic stage. Similarly, CSF analysis for depressed concentrations of amyloid and elevated concentrations of tau protein may be helpful in the preclinical stage, but superfluous in the symptomatic stage. Neurologists have not yet defined the role of PET and advanced CSF analysis in MCI.
Neurologists almost never request cerebral cortex biopsies for diagnostic purposes – mostly because routine evaluation is approximately 90% specific and sensitive.* In addition, histologic findings of Alzheimer disease differ mostly quantitatively, rather than qualitatively, from age-related changes. As a last resort, cerebral cortex biopsies can help establish a diagnosis of herpes simplex encephalitis, familial Alzheimer disease, and Creutzfeldt–Jakob disease or its variant.
Pathology
Cerebral atrophy naturally leads to compensatory dilation of the lateral and third ventricles. Of these, the temporal horns of the lateral ventricles expand the most. Nevertheless, in Alzheimer disease and most other illnesses that cause dementia, the anterior horns of the lateral ventricles maintain their concave (bowed inward) shape because of the indentation on their lateral border by the head of the preserved caudate nucleus (see Figs 20-2 and 20-18). The exception is Huntington disease, where atrophy of the head of the caudate nucleus permits the ventricle to expand laterally and assume a convex (bowed outward) shape (see Fig. 20-5).
Another histologic feature of Alzheimer disease is the loss of neurons in the frontal and temporal lobes. Neuron loss in the nucleus basalis of Meynert (also known as the substantia innominata), which is a group of large neurons located near the septal region beneath the globus pallidus (see Fig. 21-4), constitutes the most distinctive change. Their loss depletes cerebral acetylcholine (ACh: see later). Of all these histologic features, loss of neuron synapses correlates most closely with dementia.
Amyloid Deposits
Returning to the formation of Aβ, enzymes encoded on chromosome 21 cleave amyloid precursor protein (APP) to precipitate Aβ in the plaques (Fig. 7-5). Aβ differs from amyloid deposited in viscera as part of various systemic illnesses, such as multiple myeloma and amyloidosis. In Alzheimer disease, Aβ accumulates early and permanently in the cerebral cortex and vital subcortical regions.
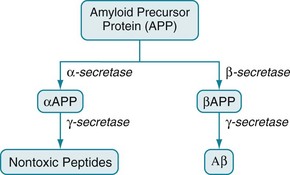
FIGURE 7-5 The amyloid precursor protein (APP) consists of 770 amino acids. Three secretase enzymes (α-, β-, and γ-) cleave it into polypeptide fragments. In one pathway, α-secretase cleaves APP to form the polypeptide αAPP. Then γ-secretase cleaves αAPP into soluble, nontoxic polypeptides. In the other pathway, β-secretase cleaves APP into a different polypeptide, βAPP. Then γ-secretase cleaves βAPP to form the insoluble, toxic, 42-amino-acid polypeptide, Aβ.
• All known genetic mutations associated with Alzheimer disease increase Aβ production (chromosomes 1, 14, 21) or Aβ aggregation (chromosome 19).
• The most powerful established genetic risk factor, which involves apolipoprotein-E (Apo-E), promotes Aβ deposition.
• Aβ is toxic in vitro probably because it binds to cerebral ACh receptors and poisons cerebral mitochondria.
• Accumulation of Aβ is a requirement for the development of Alzheimer disease.
• At least in some nonhuman studies, antibodies to Aβ reduce clinical and pathologic aspects of Alzheimer disease.
Biochemical Abnormalities
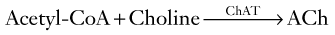
The cholinergic hypothesis, drawn from these biochemical observations, postulates that reduced cholinergic activity causes the dementia of Alzheimer disease. In addition to the histologic and biochemical data, supporting evidence includes the finding that, even in normal individuals, blocking cerebral ACh receptors causes profound memory impairments. For example, an injection of scopolamine, which has central anticholinergic activity, induces a several-minute episode of Alzheimer-like cognitive impairment that can be reversed with physostigmine (Fig. 7-6). The cholinergic hypothesis led to the introduction of donepezil (Aricept) and other cholinesterase inhibitors in an attempt at preserving ACh activity by inactivating its metabolic enzyme, cholinesterase (see Fig. 7-6 and later).
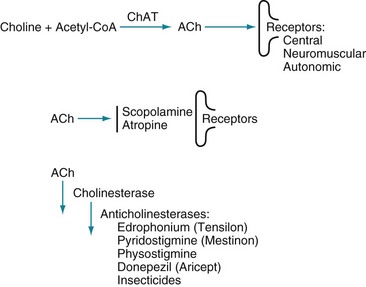
FIGURE 7-6 Top, The enzyme choline acetyltransferase (ChAT) catalyzes the reaction of choline and acetyl-coenzyme A (ACoA) to form acetylcholine (ACh) in neurons of the central (CNS), peripheral (PNS), and autonomic nervous system. When released from its presynaptic neurons, ACh interacts with postsynaptic ACh receptors. Middle, However, various substances block ACh from interacting with its receptors. For example, scopolamine, which readily crosses the blood–brain barrier, blocks ACh–receptor interaction in the CNS. Likewise, atropine blocks ACh receptors, but predominantly those in the autonomic nervous system. (Unless its concentration is great, atropine does not cross the blood–brain barrier.) Bottom, Cholinesterase metabolizes ACh. Thus, enzyme degradation rather than reuptake terminates ACh activity. (In contrast, reuptake terminates most dopamine and serotonin activity.) Anticholinesterases or cholinesterase inhibitors block cholinesterase and preserve ACh concentrations. The most widely known application of this strategy is using donepezil (Aricept) and other cholinesterase inhibitors to preserve ACh activity in Alzheimer disease. Similarly, physostigmine, a powerful anticholinesterase medicine that can cross the blood–brain barrier, purportedly briefly corrects ACh deficits in tardive dyskinesia as well as Alzheimer disease (see Chapter 18). Physostigmine might also counteract excessive anticholinergic activity in tricyclic antidepressant overdose or reverse the effects of scopolamine or atropine. Using cholinesterase inhibitors is also applicable to PNS disorders. For example, edrophonium (Tensilon) and pyridostigmine (Mestinon) are anticholinesterases that restore neuromuscular junction ACh activity in myasthenia gravis (see Fig. 6-2). In the opposite situation, insecticides contain powerful anticholinesterases that create excessive ACh activity at neuromuscular junctions, which paralyzes insects by overstimulating muscle contractions.




