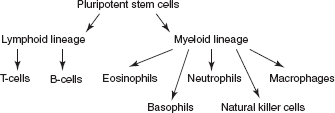
CHAPTER 13 Demyelinating Disorders I. Overview A. Commonly accepted pathologic criteria for demyelinating diseases are as follows: 1. Destruction of myelin sheaths or nerve fibers 2. Relative sparing of other elements of nervous tissue, such as axis cylinders (may be incomplete) 3. Infiltration of inflammatory cells in a perivascular distribution 4. Perivenular distribution, primarily in white matter (e.g., Dawson’s fingers at callosal septal margin due to perivenular distribution) 5. Lack of Wallerian degeneration or secondary degeneration of fiber tracts (due to integrity of the axis cylinders) B. Caveat of criteria: Schilder’s disease and necrotizing hemorrhagic leukoencephalitis may have massive damage to axis cylinders as well as myelin. C. Subacute combined degeneration, tropical spastic hemiparesis, progressive multifocal leukoencephalopathy, central pontine myelinolysis, and Marchiafava-Bignami disease were not included because of their known etiology—they are part of either viral or nutritional deficiency; metabolic deficiencies with white-matter involvement are also excluded. II. Neuroimmunology Figure 13.1 Differentiation of stem cells. A. B-lymphocytes: develop in the bone marrow; acquire immunoglobulin (Ig) receptors that commit them to a specific antigen; express IgM on the surface; after antigen challenge, T-lymphocytes assist B-lymphocytes either directly or indirectly through secretion of helper factors to differentiate and form mature antibody-secreting plasma cells. B. Igs: glycoproteins; secretory products of plasma cells; the heavy chain on Fc portion determines class: IgM, IgD, IgG, IgA, and IgE; activates complement cascade: IgM, IgG1, and IgG3. C. T-lymphocytes: thymus derived; CD4: helper cell; CD8: cytotoxic/suppressor cells; specificity of T-cells is to the foreign major histocompatibility complex (MHC) antigens; CD2 and CD3: T-cell activation. D. Natural killer cells: lymphocytes; lack immunologic memory; have the ability to kill tumor or virus-infected cells without any MHC restriction; role in tumor immunity. E. MHC and HLA: HLA lies on the short arm of chromosome 6; four major loci: class I (on all nucleated cells; HLA-A, HLA-B, HLA-C) and class II (on macrophages, B-cells, activated T-cells; HLA-DR, -DQ, -DP); class I antigens regulate the specificity of cytotoxic T-cells (CD8) and act on viruses; class II antigens regulate the specificity of helper T-cells (CD4), then CD4 regulates hypersensitivity and antibody response; examples: HLA-DR2 (multiple sclerosis [MS] among white Northern Europeans), HLA-DR3 (young myasthenic without thymoma), HLA-DR2 (narcolepsy). F. Regulation of immune response 1. Antigen cleared from immune system 2. Formation of antigen-antibody complex, which inhibits B-cell differentiation and proliferation 3. Idiotypic regulation: variable region on Ig molecule expresses proteins that are new and can act as antigens. 4. Suppressor T-cells G. Lymphokines (cytokines): secreted products of immune cells 1. Growth factors: interleukin-1, -2, -3, -4; colony-stimulating factors 2. Activation factors: interferons (α, β, and γ) 3. Lymphotoxins: tumor necrosis factors III. Multiple Sclerosis (MS) A. Also known as disseminated sclerosis, sclerose en plaques: protean clinical manifestations; usually a course of remission and relapse, but occasionally intermittently progressive or steadily progressive (especially in those >40 years old (y/o)) affecting white (more common) and gray matter and spinal cord B. Pathology: grossly numerous pink-gray (due to myelin loss) lesions scattered surrounding white matter; vary in diameter; do not extend beyond root entry zones of cranial or spinal nerves 1. Periventricular localization: characteristic, in which subependymal veins line ventricles 2. Other favored structures: optic nerves, chiasm, spinal cord; distributed randomly through brainstem, spinal cord, cerebellar peduncles 3. Astrocytic reaction: perivascular infiltration with mononuclear cells and lymphocytes; sparing of axis cylinders prevents Wallerian degeneration. Recent reevaluation of pathology shows that gray-matter pathology is common and may not extend into white matter; also, biopsy data show that this may be an early phenomenon due to pial entry of immune cells into the cortical surface. Recent pathology studies show significant axon loss even with early demyelinating events, and loss of axons even in normal-appearing white matter. C. Etiology and epidemiology: prevalence is less than 1 per 100,000 in equatorial areas, 57 to 78 cases per 100,000 in the southern United States, and 110 to 140 cases per 100,000 in the northern United States, with higher rates in Canada and Northern Europe; in southern hemisphere: less well defined; in the United States: blacks at lower risk; few “epidemics” reported. 1. Migration: before age 15 years, carries risk from native land; evidence on this is questionable, however. 2. Familial tendency also now established: 15% to 20% have an affected relative; HLA-DR2, DQW1, b1, a1, to a lesser extent -DR3, -B7, and -A3, on chromosome 6 are overrepresented in MS; low conjugal incidence (supports disease occurring early in life); first-degree relatives have a 10- to 20-fold greater risk. Monozygotic female twin risk is greater than 30%. Both parents having MS confers a 30% risk of MS to children. 3. Low incidence in children, peak at age 30 years, falling sharply in the 6th decade; two-thirds with onset between ages 20 and 40 years; may be greater in rural than urban dwellers; can occur de novo after 60, but may have had subclinical disease prior to clinical onset. 4. Popular view is that initial event is a viral infection of the nervous system with secondary activation by autoimmune reaction; role of humoral system is evident by presence of oligoclonal immune proteins in cerebrospinal fluid (CSF) that are produced by B-lymphocytes; causation not precisely known. Patients who have never had exposure to mononucleosis do not appear to get MS. 5. Cellular factor is demonstrated by abundance of helper T-cells (CD4+) in MS plaques; T-cells react to antigens presented by MHC class II on macrophages and astrocytes → stimulate T-cell proliferation, activation of B-cells, macrophages → secretion of cytokines (e.g., interferon β) → breakdown of blood–brain barrier, destruction of oligodendrocytes and myelin. 6. Physiologic effects of demyelination: impede saltatory conduction; temporary induction by heat or exercise of symptoms (Uhthoff phenomenon [visual blurring with exercise]); rise of 0.5°C can block electrical transmission; smoking, fatigue, and rise in environmental temperature all can cause worsening of symptoms. D. Clinical manifestations: weakness and numbness, both in one or more limbs, are the initial symptoms in one-half of patients; useful adage that patient with MS presents with symptoms of one leg with signs in both; Lhermitte’s phenomenon: passive flexion of the neck induces a tingling, electric-like feeling down one or more of the shoulders, arms, trunk, legs; two particular syndromes are among the most typical modes of onset. 1. Optic neuritis: in 25% of all MS patients, this is the initial manifestation; characteristically, rapid evolution over several hours to days of partial or total loss of vision, pain within the orbit, worsened by eye movement and palpation. a. Cecocentral scotoma (macular area and blind spot) can be demonstrated, as well as other field defects. b. Evidence of swelling/edema of nerve head (papillitis) in one-half of cases (distinguished from papilledema by severe vision loss). c. One-third recover completely, most improve significantly; dyschromatopsia is a frequent persistent finding; one-half or more who present with optic neuritis eventually develop MS; risk is lower in childhood. d. Uveitis and sheathing of retinal veins (due to T-cell infiltration) are other ophthalmologic findings (e.g., pars planitis) that can occur in MS. After an episode of optic neuritis, the best predictor of subsequent MS is an abnormal MRI of the brain (presence of one or more demyelinating lesions). 2. Acute transverse myelitis: transverse is imprecise: usually asymmetric and incomplete; clinically: rapidly evolving (several hours to days) paraparesis, sensory level on the trunk, sphincteric dysfunction, bilateral Babinski signs; cerebrospinal fluid (CSF): may show modest increase in lymphocytes and protein. This term can be used to refer to various disorders: a. Idiopathic severe, often following infectious or vaccination myelitis that responds incompletely to treatment with steroids or plasmapheresis, and is often monophasic b. Myelitis occurring with neuromyelitis spectrum disorders c. Myelitis with other immune conditions, such as Sjögren’s, sarcoidosis, lupus d. Partial myelitis occurring either before or during the course of MS
![]() NB:
NB:
![]() NB:
NB:
![]() NB:
NB:
Neupsy Key
Fastest Neuropsychology Insight Engine




