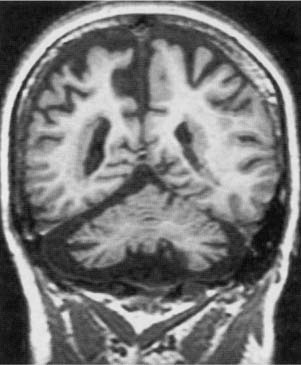
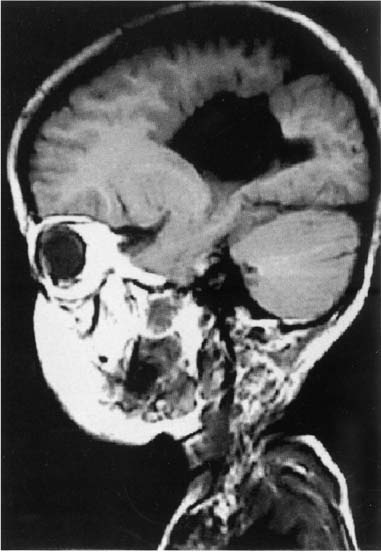
12 Note: Significant diseases are indicated in bold and syndromes in italics. 1. Cortical malformations—general symptoms include medication-refractory seizures, mental retardation, and focal neurological abnormalities a. heterotopia—formed by either the abnormal proliferation or migration of neurons; neurons are generally misshapen and incorrectly organized i. subtypes (1) focal cortical dysplasia—a gray matter connection between the cortex and the periventricular zone formed by neuroblasts that have incompletely migrated toward the cortex (2) periventricular heterotopia—clumps of neurons that failed to migrate to the cortex (or move at all) and instead reside in the periventricular white matter (Fig. 12–1) (a) some cases are related to X-linked mutations in the filamin A gene, the protein of which regulates cytoskeletal actin filament formation (3) laminar heterotopia: waves of neurons fail to migrate the entire distance to the cortex and instead form a band underneath the cortex in the white matter (Box 12.1) Laminar heterotopia and lissencephaly are both associated with mutations of the doublecortin gene (X-linked) or LIS-1 gene (autosomal dominant). b. lissencephaly—diffuse neuronal migration failure that produces only a few enlarged gyri {pachygyria} c. polymicrogyria—small cortical gyri with fusion and/or loss of the underlying neuronal layers, often occurring in a regional distribution (e.g., peri-Sylvian) d. schizencephaly—clefts in the cortex that penetrate to the ventricular system occurring in areas of polymicrogyria; the clefts are lined by cortical neurons, unlike porencephaly (Fig. 12–2) i. the severity of symptoms is related to size of the cleft and a patent cleft opening e. porencephaly—cortical clefts that can penetrate to the ventricular system, and that have openings that are surrounded by abnormal gyri (often polymicrogyria); unlike schizencephaly, clefts are usually bilateral and symmetric, and gray matter does not extend into the cleft i. clefts likely represent tissue loss from in utero injuries ii. growth of the clefts due to fluid retention may compress ventricular flow, causing hydrocephalus f. holoprosencephaly—failure of prosencephalon development, producing a smooth cortical surface without gyri or sulci i. related to (1) maternal diabetes Figure 12–1 Bilateral periventricular nodular heterotopias located on the lateral surface of the ventricles. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:73, Fig. 64. Reprinted by permission.) (2) mutation in the sonic hedgehog gene (10% of cases) (Box 12.2), which encodes a secreted intracellular signaling molecule; under expression of sonic hedgehog at the rostral end of the neural tube leads to incomplete development (3) mutations in Zic-2, a transcription regulatory protein Other mutations of the sonic hedgehog gene cause cerebellar hypoplasia and/or pituitary gland abnormalities and VACTERL syndrome. ii. subtypes (1) alobar holoprosencephaly: no divisions develop between the hemispheres or lobes; associated with a single underlying ventricle, agenesis of the septum and corpus callosum, and olfactory and optic nerve hypoplasia (2) semilobar prosencephaly: failure of the anterior interhemispheric division allows for fusion of the anterior cortices with significant hypoplasia of the corpus callosum and olfactory nerves; a posterior interhemispheric division exists and the basic lobar structure is otherwise preserved (a) septo-optic dysplasia—a variant of semilobar prosencephaly; specific features include hypothalamic hamartomas causing panhypopituitarism, agenesis of the cerebellar vermis and fusion of the dentate nuclei causing ataxia (Fig. 12–3). (3) lobar prosencephaly: interhemispheric connections occurring between frontal poles and the cingulate cortices through an oversized indusium griseum (a vestigial gray matter band normally located on top of the corpus callosum); limited hypoplasia of the corpus callosum g. hydranencephaly—reduction of the cerebral cortex to a flat, thin layer, often sparing the basal cortex and hippocampus but severely disrupting the thalamus and basal ganglia; caused by destructive processes (which produce microcephaly at birth) or by hydrocephalus (which produces macrocephaly at birth) Figure 12–2 Schizencephaly. (From Citow JS et al. Neuropathology and Neuroradiology: A Review. Thieme; Stuttgart, Germany: Georg Thieme; 2001:11, Fig. 9. Reprinted by permission.) Figure 12–3 Septo-optic dysplasia with absent septum pellucidum between the two frontal horns (A) and thin optic nerves (B). (From Citow JS et al. Neuropathology and Neuroradiology: A Review. Thieme; Stuttgart, Germany: Georg Thieme; 2001:10, Fig. 7A. Reprinted by permission.) i. generally fatal within a few days due to hypothalamic dysfunction 2. Subcortical malformations a. agenesis of the corpus callosum—caused by complete or partial failure of the extension of the developing corpus callosum posteriorly from the lamina terminalis; fibers that cannot cross in the corpus callosum run ipsilaterally in a rostrocaudal direction {bundle of Probst} (Box 12.3) Aicardi’s syndrome Pathophysiology—X-linked inherited agenesis of corpus callosum and anterior commissure, and polymicrogyria Symptoms—Mental retardation; myoclonic epilepsy; chorioretinal abnormalities; vertebral abnormalities i. other commissures are enlarged ii. occurs in isolation in 2% of population iii. symptoms: generally asymptomatic without other malformations, but may exhibit (1) learning disabilities or mental retardation (2) abnormally wide-spaced eyes {hypertelorism} (3) exotropia with inability to converge 3. Abnormalities of cerebrospinal fluid and the ventricular system a. symptoms of hydrocephalus include irritability, bulging fontanelles, prominent scalp veins, false-localizing CN VI palsies, Parinaud’s syndrome, hyperreflexivity, and irregular respiration b. diagnosis requires a head circumference > 2 standard deviations for gestational age or an increase in head circumference > 2 standard deviations during first year of life; diagnosis does not require elevated intracranial pressure, which only occurs after fusion of the cranial sutures c. pathophysiology: acquired pediatric hydrocephalus conditions i. noncommunicating: aqueduct stenosis caused by intrauterine infection, hemorrhage, trauma, or tumor ii. communicating: following subarachnoid hemorrhage or meningitis d. pathophysiology: syndromal hydrocephalus conditions i. trisomy 9, 13, 18 ii. VACTERL syndrome with hydrocephalus (Box 12.4) VACTERL syndrome Vertebral anomalies; anal atresia; cardiac defects; tracheo esophageal fistula; renal abnormalities; limb abnormalities. Some cases can be caused by mutation in the sonic hedgehog gene, like holoprosencephaly. iii. Hunter’s and Hurler’s syndromes iv. Walker-Warburg syndrome (also has congenital muscular dystrophy, lissencephaly, Dandy-Walker malformation, and eye malformations) v. craniosynostosis syndromes: all involve premature closure of skull sutures leading to skull deformation, and have autosomal dominant and sporadic forms (1) simple craniosynostosis: early closure of one or a few sutures that does not cause hydrocephalus or other symptoms (2) complex craniosynostosis (a) Crouzon’s syndrome—other symptoms include multiple cranial nerve palsies caused by jugular foramen narrowing; no mental retardation (b) Apert’s syndrome—other symptoms include facial deformity, syndactyly, short upper extremities, and mental retardation vi. X-linked hydrocephalus syndromes: both are caused by mutations in L1CAM, a cell adhesion molecule (Box 12.5) (1) mental retardation, aphasia, shuffling gait, adducted thumbs (MASA) disorder (2) X-linked aqueduct stenosis—also exhibits adducted thumbs, agenesis of the corpus callosum, brainstem malformations, and hypoplastic corticospinal tracts 4. Cerebellar and brainstem malformations a. Chiari malformations
Developmental and Metabolic Diseases of the Nervous System
I. Disorders of Embryogenesis
A. Malformations
Box 12.1
Box 12.2
Box 12.3
Box 12.4
| Chiari I malformation | Chiari II malformation |
Symptoms | Onset in young adulthood | Onset in infancy |
| Head, neck, or upper back pain; extremity spastic weakness ± distal atrophy; extremity sensory loss; gait ataxia; diplopia; downbeat nystagmus | Dysphagia: cyanosis during feeding, regurgitation, aspiration; apnea, stridor; upper extremity weakness; opisthotonos; nystagmus, particularly downbeat |
Cerebellum | Downward herniation of cerebellar tonsils; elongated, peg-like tonsils | Downward herniation of cerebellar vermis; dysplastic, hypomyelinated cerebellar folia |
Brainstem | Normal | Herniation of medulla into the spinal canal; buckling of medulla {Z deformity}, tectal beaking; stretched and atrophic lower cranial nerves |
Spine and spinal cord | Syringomyelia, scoliosis | Myelomeningocele; syringomyelia, scoliosis |
Ventricular system | Hydrocephalus due to adhesions occluding 4th ventricle foramen (< 5%) | Hydrocephalus from cerebral aqueduct stenosis; big occipital horns of lateral ventricles. {colpocephaly} |
Skull | Minor abnormalities of basilar skull | Enlarged foramen magnum; multifocal skull defects {luckenschadel} (See figure at right.); platybasia/basilar impression |
Supratentorial structures | Normal | Hypoplastic and irregular tentorium and falx {Chinese lettering sign}; enlarged thalamic massa intermedia, absent septum; agenesis of corpus callosum |
Diagnostic testing | Neuroimaging: Descent of one tonsil > 5 mm below foramen magnum, or both tonsils 3–5 mm below foramen magnum | Neuroimaging: For aforementioned abnormalities; skull films demonstrate luckenschadel |
Treatment and prognosis | Craniocervical decompression | May need ventricular shunting |
|
| Craniocervical decompression |
| Good outcome with pain or cerebellar injury; poor outcome with central cord injury | Older patients (i.e., children) tend to improve after surgery; unclear if decompression alters course in infants |
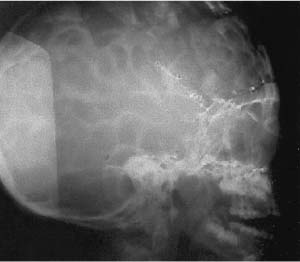
Luckenschadel. (From Citow JS et al. Neuropathology and Neuroradiology: A Review. Thieme; Stuttgart, Germany: Georg Thieme; 2001:14, Fig. 11, Fig. 13A. Reprinted by permission.)
i. Chiari I and II malformations (Table 12–1) (Fig. 12–4)
ii. other Chiari malformations
(1) Chiari III malformation: a very rare malformation characterized by herniation of an abnormal brain stem and cerebellum into a low occipital/high cervical encephalocele; associated with hydrocephalus
(2) Chiari IV malformation: aplasia or hypoplasia of the cerebellum, therefore not a true Chiari malformation due to the lack of any hindbrain herniation
(3) Chiari 0 malformation: syringohydromyelia not associated with hindbrain herniation or other apparent etiology, likely due to functional disturbances of cerebrospinal fluid flow at the foramen magnum
b. Dandy-Walker malformation (Box 12.6)
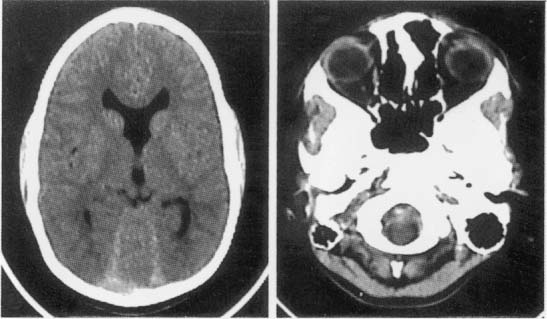
i. pathophysiology: atresia of the foramen of Luschka and Magendie together with agenesis of the cerebellar vermis creates a fluid space that communicates with the subarachnoid space (Fig. 12–5)
Box 12.6
Dandy-Walker malformations look similar to posterior fossa subarachnoid cysts, which do not communicate with the subarachnoid space as does the Dandy-Walker malformation.
(1) associated abnormalities include agenesis of the corpus callosum (20%) and occipital encephalocele (10%); rarely associated with true hydrocephalus
(a) may be associated with cleft palate, ocular abnormalities (iris defects {coloboma}, microphthalmia), and cardiac structural defects
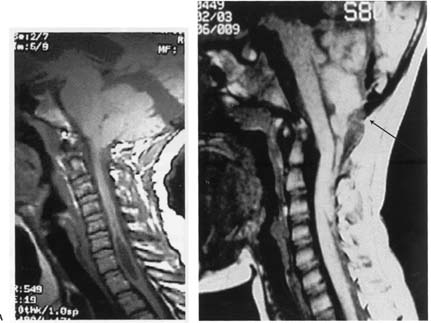
Figure 12–4 Chiari I malformation with syrinx (A), Chiari II malformation with mesencephalic malformation (“tectal beaking”), low-lying torcula (arrow), medullary/cervical spinal cord buckling, and descent of the cerebellar vermis. (From Citow JS et al. Neuropathology and Neuroradiology: A Review. Thieme; Stuttgart, Germany: Georg Thieme; 2001:13, Fig. 11, Fig. 12. Reprinted by permission.)
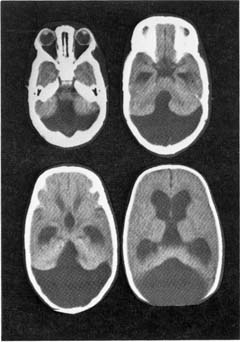
Figure 12–5 Dandy-Walker malformation demonstrating a hypoplastic cerebellar vermis, laterally displaced cerebellar hemispheres, retrocerebellar cyst, and hydrocephalus. (From Mori K. Anomalies of the Central Nervous System. Thieme; Stuttgart, Germany: Georg Thieme; 1985:92, Fig. 6–12A. Reprinted by permission.)
ii. symptoms: mental retardation; ataxia; spasticity; seizures (15%)
iii. treatment: if symptomatic, shunt the posterior fossa fluid space; ventricular shunts in absence of posterior fossa shunt may cause upward herniation
5. Spinal malformations
a. spina bifida: a neurulation abnormality
i. subtypes: all are most common in lumbosacral region and may be associated with an overlying tuft of hair
(1) spina bifida occulta: incidental defect in the dorsal bony vertebra; occurs in 20% of the general population
(2) meningocele: a meningeal outpouching that does not contain neural tissues and is well-covered by skin; has minimal association with brain malformations
(a) treatment: early surgical closure
(3) myelomeningocele: failure of the posterior neuropore closure; meningeal outpouching includes neural tissues and is covered by minimal skin elements (Box 12.7)
Box 12.7
Other Neurulation Abnormalities
Anencephaly—Failure of anterior neuro-pore closure; extends caudally as failure of spinal cord neurulation {rachischisis} Encephalocele—Unilateral cortical protrusion through a midline skull defect (occipital > frontal)
(a) always associated with a type II Chiari malformation, and may have hydrocephalus from aqueduct stenosis (80%)
(b) treatment: early surgical closure, although this is unlikely to improve the patient’s quality of life
ii. diagnostic testing: prenatal diagnosis with elevated amniotic fluid α-fetoprotein and ultrasound
iii. treatment: prenatal folate supplementation reduces the incidence of spina bifida
b. tethered spinal cord
i. pathophysiology: stretching of the spinal cord by an unusually strong filum terminalis or lipoma, causing ischemia; associated with spina bifida occulta in 90% of cases, and the tethered cord usually accounts for deterioration in such patients who have back pain (deterioration without back pain is usually due to syringomyelia)
(1) can be associated with chromosomal abnormalities (trisomy 13, 18; DiGeorge syndrome)
ii. symptoms: back pain; lower extremity weakness and sensory loss; lower extremity atrophy and/or foot deformities; bowel/bladder dys-function; progressive kyphoscoliosis; delayed walking; constipation (most common presenting sign in children); imperforate anus and other anal anomalies
iii. treatment: surgical transection of the filum; removal of any lipoma
c. caudal regression syndrome/sacral agenesis
i. pathophysiology: degeneration of the coccyx and sacral vertebrae, and of the sacral spinal cord that causes atrophy of the lower extremities and malformations of the rectum and genitourinary system; caused by decreased expression of the sonic hedgehog protein at the caudal end of neural tube
(1) also associated with maternal diabetes mellitus
6. Vein of Galen malformations: Caused by direct arterial connections into the vein of Galen
a. subtypes
i. neonatal onset: the shunt involves a significant proportion of the cardiac output, therefore it presents as high-output congestive heart failure
ii. infantile onset: presents with hydrocephalus, likely due to obstruction of the aqueduct of Sylvius by the enlarged vein of Galen
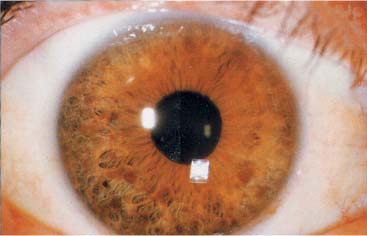
Figure 12–6 Lisch nodules. (From McKhann GM et al. Q&A Color Review of Clinical Neurology and Neurosurgery. Stuttgart, Germany: Georg Thieme; 2003:93, Fig. 87a. Reprinted by permission.)
iv. adult onset: presents as hemorrhagic infarction
b. treatment: endovascular occlusion of the arterial connections
II. Neurocutaneous Syndromes/Phakomatoses
A. Neurofibromatosis Type 1/von Recklinghausen’s Disease
1. Pathophysiology: caused by mutation of NF-1 gene on chromosome 17, which encodes neurofibromin protein that is likely a GTPase-activating protein; somatic mosaicism may cause localization of abnormalities to one part of the body
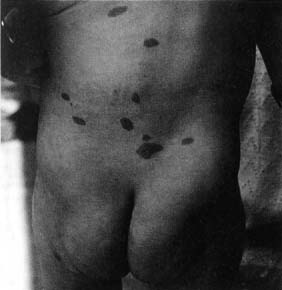
Figure 12–7 Café-au-lait spots. (From Panteliadis CP, Darras BT. Encyclopaedia of Paediatric Neurology: Theory and Practice, 2nd ed. Stuttgart, Germany: Georg Thieme; 1999:491, Fig. 25–1. Reprinted by permission.)
a. histology
i. subcutaneous neurofibromas develop along peripheral nerves; neurofibromas and/or schwannomas can develop on cranial nerves, but not as frequently as in neurofibromatosis type 2
ii. gliomas (usually juvenile pilocytic astrocytomas) develop on the optic nerve (25%) of in the brain parenchyma (5%)
iii. pigmented hamartomas or the iris {Lisch nodules (Fig. 12–6)} or retina
2. Symptoms: onset by 5 years of age (Box 12.8)
Box 12.8
Diagnostic Criteria for Neurofibromatosis (NF) Type 1
Requires two or more of the following:
 At least six café-au-lait spots that are > 5 mm before puberty or > 15 mm after puberty
At least six café-au-lait spots that are > 5 mm before puberty or > 15 mm after puberty
 Multiple neurofibromas or one plexi-form neurofibroma
Multiple neurofibromas or one plexi-form neurofibroma
 Multiple Lisch nodules
Multiple Lisch nodules
 Axillary freckling
Axillary freckling
 Optic glioma
Optic glioma
 Bony lesions: thinning of long bones, pseudoarthrosis
Bony lesions: thinning of long bones, pseudoarthrosis
 A family history of NF type 1
A family history of NF type 1
a. neurological symptoms
i. mental retardation (40%), seizures (10%)
ii. painful peripheral neuropathies or radiculopathies from subcutaneous neurofibromas
b. eye: abnormalities are asymptomatic
c. skin: café-au-lait spots (Fig. 12–7); axillary freckling
d. hypertension from pheochromocytoma and/or renal artery stenosis
3. Diagnostic testing
a. neuroimaging: MRI demonstrates areas of non-enhancing increased T2 and diffusion-weighted signal {unidentified bright objects (UBOs)} that are located commonly in the basal ganglia, brainstem, and cerebellum (Fig. 12–8)
i. UBOs likely represent areas of glial proliferation, and they are frequently sites for glioma development; 40% will spontaneous resolve, but persistence of UBOs into adulthood is associated with mental retardation
b. genetic abnormalities of NF-1 gene are detected in only 70%
4. Treatment: None specific
B. Neurofibromatosis Type 2
1. Pathophysiology: mutation of NF-2 gene on chromosome 22, encodes merlin/schwannomin protein, which acts as a tumor suppressor (Box 12.9)
a. histology: multiple schwannomas, classically bilateral on CN/VIII but also on peripheral nerves; frequently associated with ependymomas > meningiomas and astrocytomas
2. Symptoms: typically develop around 20 years of age (Box 12.10)
Box 12.10
Diagnostic Criteria for Neurofibromatosis Type 2: Requires All Three of the Following
 Bilateral CN VIII tumors
Bilateral CN VIII tumors
 Family history of neurofibromatosis type 2
Family history of neurofibromatosis type 2
 One family member with a CN VIII tumor or two other nervous system tumors (neurofibroma, schwannoma, meningioma, glioma)
One family member with a CN VIII tumor or two other nervous system tumors (neurofibroma, schwannoma, meningioma, glioma)
a. neurological symptoms: vertigo, hearing loss, tinnitus from CN/VIII schwannomas (usually develop on the vestibular portion); schwannomas > neurofibromas on peripheral nerves may cause painful peripheral neuropathies
i. may develop facial weakness from compression of CN/VII in the internal acoustic meatus
b. skin: as per neurofibromatosis type 1 but less prominent; café-au-lait spots and subcutaneous neurofibromas are rare
c. eye: rare Lisch nodule
3. Diagnostic testing: neuroimaging with MRI; genetic testing
4. Treatment: none specific
C. Tuberous Sclerosis
1. Pathophysiology
a. caused by autosomal dominant mutations in the
i. TSC-1 gene (chromosome 9): encodes the hamartin protein, which functions in linking the cell membrane to the cytoskeleton
ii. TSC-2 gene (chromosome 16): encodes the tuberin protein, which is similar to GTPase-activating proteins and interacts with hamartin
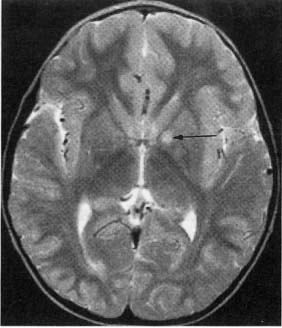
Figure 12–8 Unidentified bright objects (arrow) of neurofibromatosis type 1. (From Raininko R et al. Non-neoplastic brain abnormalities on MRI in children and adolescents with neurofibromatosis type 1. Neuropediatrics, 2001, 32: 226, Fig. 1b. Reprinted by permission.)
b. TCS1 mutations may be more likely to cause familial disease and have a milder course, but otherwise there is no obvious phenotype difference between the two genetic subtypes
2. Histology
a. nervous system: cortical hamartomas {tubers}; focal cortical hypoplasia; subependymal heterotopias that are generally calcified; subependymal giant cell astrocytomas
b. eye: hamartomas, retinal depigmentation
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


