(1)
Cognitive Function Clinic, Walton Centre for Neurology and Neurosurgery, Liverpool, UK
8.3 Dementia with Lewy Bodies, Parkinson’s Disease Dementia, and Other Parkinsonian Disorders (PSP, CBD)
8.5 Prion Disease
Abstract
This chapter examines the various cognitive disorders (e.g. Alzheimer’s disease, frontotemporal lobar degenerations, parkinsonian disorders) which may be defined by clinical assessment and investigation, emphasizing their clinical heterogeneity.
Keywords
DementiaDiagnosisCognitive disordersAlzheimer’s diseaseFrontotemporal lobar degenerationParkinsonian disordersVascular dementiaThe delineation of cognitive syndromes (see Chap. 7) may narrow differential diagnostic considerations for specific dementia disorders. There are many causes of the dementia syndrome and of cognitive impairment (Mendez and Cummings 2003; Kurlan 2006; Larner 2008a, 2010a, 2013a; 2014; Filley 2012). Only those most often encountered in CFC practice are discussed briefly here; current diagnostic criteria are listed in Chap. 2 (Box 2.7). Other disorders, such as delirium (Sect. 7.2.2) and depression (Sect. 5.2.2), may need to be considered in the initial differential diagnosis, as independent or superimposed causes of cognitive impairment.
8.1 Alzheimer’s Disease
Alzheimer’s disease (AD) is by far the most common neurodegenerative disorder seen in CFC, the relatively young age of the casemix notwithstanding. Around 95 % of these AD cases have presented with the typical amnesic syndrome, the remainder comprising the focal cortical variants presenting with agnosia, aphasia, apraxia, or dysexecutive syndrome (Larner 2006a, 2008b). These variant presentations (Caselli and Tariot 2010) are acknowledged in updated AD diagnostic criteria (McKhann et al. 2011).
Errors in the diagnosis of AD sometimes occur, even in the best centres. We have encountered patients diagnosed with AD elsewhere who have proved not to have dementia on longitudinal follow up. Over-reliance on structural brain imaging reported to show “atrophy” may have been instrumental in the misdiagnosis of these cases (see Sect. 6.2.1; Larner 2004).
An examination of the Cognitive Function Clinic (CFC) database of patients in whom either an initial diagnosis of AD was made and/or cholinesterase inhibitors were prescribed, covering the period January 2000 to end June 2008 (8½ years), was interrogated to identify those in whom diagnostic revision was required, based on disease progression with emergence of new features during follow-up (Davies and Larner 2009). Of 300 patients on the database, 289 (= 96.3 %) were initially clinically diagnosed with probable AD using NINCDS-ADRDA criteria (McKhann et al. 1984). From this group, 8 patients initially diagnosed with AD in whom subsequent diagnostic revision was required were identified (= 2.8 %; M:F = 7:1, age range at diagnosis 52–63 years, median 58 years). In all cases, diagnosis was revised from AD to FTLD due to the emergence, in isolation or combination, of behavioural (7), linguistic (2), and motor (1) features more typical of the FTLD phenotypes. Onset of these changes was noted between 12 months and 5 years (median 18 months to 2 years) after initial diagnosis. Two patients were eventually shown to harbour tau gene mutations (Larner 2008c, 2009a); both had a family history of early-onset dementia (parent or siblings affected), but in neither case did the available details permit the conclusion of autosomal dominant inheritance of disease (i.e. ≥3 affected in individuals in 2 generations; Cruts et al. 1998). One of these patients developed prototypical FTLD behavioural features, the other a phenotype of progressive supranuclear palsy, 3 and 4 years after initial diagnosis respectively. Of the six other patients, all with early-onset disease (i.e. onset ≤ 65 years of age), none had a family history of dementia and hence all were initially diagnosed with sporadic probable AD. Two were initially thought to have aphasic presentations of AD, since they had apparent amnesia in addition to aphasia, but both gradually developed behavioural features requiring their reclassification as FTLD. A further two patients also evolved behavioural features after amnesic presentations. The two remaining patients with amnesic presentations developed progressive impoverishment of language function suggestive of the progressive non-fluent aphasia phenotype, as well as behavioural features. In one of these cases, English was not the patient’s first language, thereby confounding initial assessment. That FTLD may on occasion be misdiagnosed as AD is perhaps not surprising, as there is symptom overlap between AD and FTLD as defined by widely accepted clinical diagnostic criteria (Varma et al. 1999). In clinical practice initial assessment is essentially cross-sectional, whilst longitudinal assessment may reveal new features mandating diagnostic revision. The adoption of more modern diagnostic criteria for AD (Dubois et al. 2007; McKhann et al. 2011) and FTLDs (Gorno-Tempini et al. 2011; Rascovsky et al. 2011) may obviate this problem in the future.
Genetically determined cases of AD have been rarely encountered (see Sect. 6.3.1), all those seen having presenilin 1 gene mutations (Larner and du Plessis 2003; Doran and Larner 2004a, 2006; Larner et al. 2007). Patients with Down syndrome invariably harbour AD pathology after the age of 50 years (Mrak and Griffin 2004), presumably because of the extra copy of the APP gene in trisomy 21. The neuropathological features may have the clinical correlate of cognitive decline and dementia, but such cases have rarely been seen in CFC (see Sect. 8.6; Larner 2007, 2011a).
8.2 Frontotemporal Lobar Degeneration
Frontotemporal lobar degenerations (FTLD) are less common than AD overall but in the presenile age group they may be as prevalent as AD (Ratnavalli et al. 2002), although other population based studies find AD to be more prevalent in this age group (Harvey et al. 2003). Hence FTLDs make up a significant component of CFC work, in part because of the relatively young age at onset of patients seen in this setting (see Sect. 1.3). In addition, patients referred to CFC from psychiatrists (see Sect. 1.2.2) have an increased frequency of FTLD, mostly the behavioural variant. FTLD in the elderly may be underreported, and may differ in clinical and pathological phenotype from early-onset disease (Baborie et al. 2012, 2013).
FTLDs are heterogeneous at the clinical, neuropsychological, neuropathological and genetic level (Hodges 2007; Seelaar et al. 2011). Of the various clinical phenotypes encompassed by the FTLD rubric (Neary et al. 1998), behavioural variant (bvFTD; Rascovsky et al. 2011) and the agrammatic variant of primary progressive aphasia (avPPA; formerly known as progressive nonfluent aphasia) are more common than semantic dementia (Gorno-Tempini et al. 2011), and this has been the experience in CFC (Larner et al. 2005a; Davies and Larner 2010; Larner 2012a), consistent with reports from other centres seeing larger numbers of FTLD cases (Case Studies 4.3 and 6.3).
Cases of FTLD with motor neurone disease (FTD/MND) have also been seen, many referred from psychiatry clinics or under concurrent care of psychiatrists (see Sect. 1.2.2; Doran et al. 2005; Hancock and Larner 2008; Larner 2008d, 2013b; Sathasivam et al. 2008; Larner and Gardner-Thorpe 2012). It is of note that FTD/MND is not mentioned in DSM-IV-TR (American Psychiatric Association 2000), either as a specific cause of dementia or in the catch-all category of “Dementia due to other general medical conditions”. This omission is surprising in light of the fact that FTD/MND may present with neuropsychiatric symptoms (Sect. 7.2.1), leading to referral to psychiatrists rather than neurologists in the first instance. These neuropsychiatric symptoms include disinhibition, which may be mistaken for hypomania, and self-neglect and poverty of speech which may be mistaken for depression (Sathasivam et al. 2008), as well as florid delusions. Disinhibition was presumably the substrate for the “animal-like behaviour” seen in one patient with bvFTD who, according to his wife, used to bark like a dog, a behaviour which may fall under the rubric of lycanthropy (Larner 2010b).
As far as genetically determined cases of FTLD are concerned (Sect. 6.3.2), it was the case that large centres reported either a preponderance of tau compared to progranulin mutations (Seelaar et al. 2008) or roughly equal numbers (Rohrer et al. 2009), but since the discovery of the C9orf72 hexanucleotide repeat mutation this has superseded both tau and progranulin in frequency (Boeve et al. 2012; Dobson-Stone et al. 2012; Hsiung et al. 2012; Mahoney et al. 2012; Simon-Sanchez et al. 2012; Snowden et al. 2012).
Families with tau gene mutations (FTDP-17) have been seen on occasion in CFC (see Sect. 6.3.2). In some of these cases the proband received an initial diagnosis of probable AD, with features more typical of FTLD only emerging at a later stage of disease. This clinical heterogeneity has also been observed with some of the other tau gene mutations (Larner and Doran 2009a), such as R406W (Lindquist et al. 2008). This diagnostic error, FTLD confused with AD, has also been noted in sporadic FTLD patients (Davies and Larner 2009) perhaps related to the overlap of older diagnostic criteria (Varma et al. 1999). Occasional FTLD cases with progranulin and C9orf72 mutations have also been seen (Sect. 6.3.2; Larner 2012b, 2013b; Ziso et al. 2014).
Unusual forms of FTLD have also been seen, defined on neuropathological grounds. Neuronal intermediate filament inclusion disease (NIFID) was initially defined by intraneuronal cytoplasmic inclusions of variable morphology which immunostained for all class IV intermediate filament (IF) proteins, namely NF-H, NF-M, NF-L, and alpha-internexin (Cairns et al. 2004). More recently it has been shown that a much larger proportion of the inclusions in NIFID are immunoreactive with the fused in sarcoma (FUS) protein than with IF (Neumann et al. 2009), leading to changes in the suggested nomenclature to FTLD-FUS (Mackenzie et al. 2010). These cases have a broad phenotype which may overlap with both corticobasal degeneration and motor neurone disease, and the pathological diagnosis may be unsuspected ante mortem (Menon et al. 2011).
Late diagnosis of FTLD is a common problem, even following contact with medical services, with an average delay of nearly 3 years in a Scandinavian series in which nearly three-quarters of patients initially received a non-dementia diagnosis (Rosness et al. 2008). Such delays are of particular frustration to caregivers who are often sure something is wrong. An integrated care pathway (ICP) has been developed in the hope of hastening FTLD diagnosis (see Sect. 9.6; Davies and Larner 2010).
8.3 Dementia with Lewy Bodies, Parkinson’s Disease Dementia, and Other Parkinsonian Disorders (PSP, CBD)
Dementia with Lewy bodies (DLB) is claimed to be the second most common of the neurodegenerative dementias by some authors, but has been encountered relatively rarely in CFC, in contrast to other centres, although this low prevalence does appear to be within the range of prevalence estimates for the general population (Zaccai et al. 2005). Presence of REM sleep behaviour disorder (REMBD), sometimes referred to as “dream enactment”, may be a useful clue to diagnosis, and is often amenable to treatment with clonazepam (Larner et al. 2005b). Based on the greater impairment of attentional and visuospatial function, and the relative preservation of orientation and memory function, in DLB as compared to AD (e.g. Salmon et al. 1996; Downes et al. 1998; Ballard et al. 1999; Calderon et al. 2001), Ala et al. (2002) derived a weighted subscore from the Mini-Mental State Examination (MMSE) for DLB diagnosis. Prospective use of the Ala subscore (and its modifications derived from the ACE and MoCA) has not proved of particular use in CFC for prospective diagnosis (see Sects. 4.1.1, 4.5.1, and 4.9.1).
DLB may sometimes be mistaken for CJD (e.g. Haik et al. 2000; Tschampa et al. 2001; Van Everbroeck et al. 2004; Larner 2006b; Du Plessis and Larner 2008), not least because rapidly progressive instances of DLB have been described (Momjian-Mayor et al. 2006; Gaig et al. 2011). One differential diagnostic clue is that the visual hallucinations of DLB are generally well formed (animals, people) compared with the rather elemental visual hallucinations (colours, shapes) which may occur in CJD. EEG findings of periodic sharp wave complexes may sometimes be found in DLB, adding to the phenotypic overlap (see Sect. 6.4.1; Doran and Larner 2004b). Orthostatic hypotension may be a feature, sometimes initially in isolation prompting diagnosis of pure autonomic failure (Larner et al. 2000). This may predispose to repeated syncope, one of the supporting features in DLB diagnostic criteria (McKeith et al. 2005).
Parkinson’s disease dementia (PDD) is likely to become an increasing problem, since most patients with PD followed longitudinally develop some evidence of cognitive decline over time (Reid et al. 2011; Williams-Gray et al. 2012). Few patients with PDD have been seen in CFC presumably because they are managed in either dedicated movement disorder clinics or, because of the neuropsychiatric problems, psychiatry clinics. Instruments such as the MMP and MoCA (see Sects. 4.2 and 4.9) may be useful for the detection of cognitive impairments in PDD.
Parkinsonian syndromes other than PDD and DLB may be accompanied by neuropsychological impairment as well as movement disorder (Larner 2013a:48–51), in particular progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD). Occasional cases of PSP have been seen in CFC; the phenotype has also been seen in association with tau gene mutations (Larner 2009a, 2012c; Larner and Doran 2009a) and in a case of Perry syndrome (Aji et al. 2013), as well as being mistaken for normal pressure hydrocephalus (Schott et al. 2007). Cases of suspected CBD but with other pathological substrates, so called corticobasal syndrome (Boeve et al. 2003; Doran et al. 2003), are well-recognised.
8.4 Vascular Dementia, Vascular Cognitive Impairment
Vascular dementia (VaD) and vascular cognitive impairment (VCI) are recognised to be heterogeneous entities with respect to both pathology and pathogenesis (Wahlund et al. 2009; Gorelick et al. 2011), including vasculopathic and thrombotic disorders. Mixed dementia, defined as the coexistence of AD and VaD (Langa et al. 2004), may be the most common neuropathological substrate of dementia (MRC CFAS 2001; Schneider et al. 2009). Cerebrovascular disease may modulate the clinical expression of AD pathology (Snowdon et al. 1997). The old dichotomy of AD and VaD is now superseded by an integrative approach to aetiology with a continuum or spectrum running from pure boundary cases through entities such as “AD with vascular lesions” and “VaD with AD changes”.
Cases of pure vascular dementia, such as subcortical ischaemic vascular dementia (Román et al. 2002), have rarely been encountered in CFC. Two cases with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) have been seen (see Sect. 6.3.3). Though unusual, dural arteriovenous fistula (dAVF) must also be considered amongst reversible causes of vascular cognitive impairment and dementia (Fig. 8.1). Experience with three cases of intracranial dAVF seen at CFC (Wilson et al. 2010) found impairments in attention, memory and executive functioning in all cases. One common clinical feature which was not fully captured by the standard neuropsychological and cognitive tests administered was the impairment in processing speed, suggestive of subcortical involvement. This may have been a reflection of the marked prolongation of cerebral transit time seen with radiological contrast studies, late angiographic views indicating that venous drainage of brain parenchyma was considerably delayed. Of note, despite marked cognitive improvement after endovascular fistula embolisation, residual deficits were evident in some cognitive domains even up to 2 years after treatment, presumably related to irreversible structural changes in the brain, such as complete or partial venous infarction of tissues subjected to chronic venous hypertension.
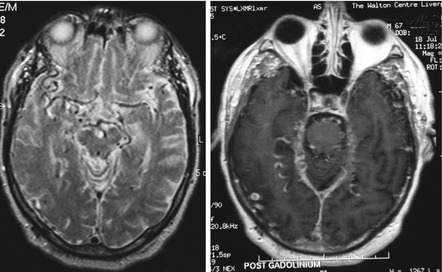
Fig. 8.1
MRI brain scan showing abnormally dilated vessels on T2 weighted (left) and post contrast T1 weighted (right) sequences, suggestive of dural arteriovenous fistula (Wilson et al. 2010)
8.5 Prion Disease
Prion diseases have attracted much attention in recent years, not least because of their novel biology as sporadic, inherited, and iatrogenic conditions (Collinge 2001), and despite their clinical rarity.
An audit of prion cases seen at CFC over a 12-year period (1990–2001 inclusive) (Larner and Doran 2004) found that 82 patients with suspected CJD were referred from the Mersey Region to the National Creutzfeldt-Jakob Disease Surveillance Unit (NCJDSU) in Edinburgh; 65 referrals were made after 1995 when the UK epidemic of variant CJD (vCJD) began (Will et al. 1996). Sixty-six patients (80 %) presented initially to non-neurologists. Forty-four referrals were of inpatients at WCNN, usually transferred from district general hospitals by visiting neurologists. Thirty-eight cases were referred to NCJDSU directly from district general hospitals or from Alder Hey Children’s Hospital, Liverpool. Prion disease was confirmed pathologically in 43 of 82 referrals, giving an overall diagnostic accuracy of 0.52. Of the confirmed prion disease cases, 33 had sporadic CJD, 8 had vCJD (e.g. Silverdale et al. 2000; Lorains et al. 2001), and 2 had iatrogenic disease; there were no familial cases. Of the non-prion cases (39), eight were found to have alternative diagnoses only at postmortem, principally AD and DLB (see Sects. 8.1 and 8.3). Autoimmune encephalopathy may also mimic CJD (Schott et al. 2003; Geschwind et al. 2008).
Although diagnosis of prion disease may be straightforward (Case Study 8.1), there may be difficulties if the phenotype is unusual, for example with prominent parkinsonism and orthostatic hypotension (Du Plessis and Larner 2008), or there is a long prodrome of psychiatric symptoms (Ali et al. 2013). Neuropsychiatric features, once claimed to be a distinguishing feature of vCJD, are in fact quite common in sCJD, even early in the disease course (Wall et al. 2005; Rabinovici et al. 2006). They were also prominent in another patient seen in CFC whose non-identical twin was discordant for the disease. Whether subclinical vCJD, which may be more common than previously thought (Gill et al. 2013), might manifest with different clinical features, particularly in patients valine homozygous at PRNP gene codon 129, remains to be seen.
Case Study 8.1: Clinical Diagnosis: Sporadic CJD
A 75 year-old lady was brought to CFC by ward staff from another hospital; she was unable to give any history. Previously very fit and active, she had apparently developed cognitive problems over a 5-month period. A month or so after symptom onset her MMSE was 21/30 and a CT brain scan was reported to be normal. However her decline was relentless, requiring hospital admission because of failure to cope at home. Aside from some myoclonic jerks her neurological examination was normal. A diagnosis of sporadic CJD was suspected on the basis of the rapid decline and the myoclonic jerks. Subsequent EEG was abnormal with a non-specific slow background but no triphasic waves were seen. CSF analysis was positive for 14-3-3 protein.
8.6 Learning Disability; Down Syndrome
The assessment of individuals with learning disability remains problematic for most neurologists, since generally they have received little or no training in this area, far less developed any claims to expertise. Most patients with learning disability are referred to neurology services because of episodes of loss of or impaired consciousness which may reflect epileptic seizures (see Sect. 7.2.3) (Adab and Larner 2006; Larner 2007, 2009b, 2011a; Sells and Larner 2011), although occasional patients are sent to CFC with possible progression of cognitive dysfunction. Cases of learning disability in the context of neurofibromatosis-1 and fragile X syndrome have sometimes been seen (Larner 2008e).
Many forms of learning disability are inadequately understood at the pathological or aetiological level, but some are better characterised. For example, in those with Down syndrome (trisomy 21), cognitive decline often reflects the inevitable development of Alzheimer pathology (Mrak and Griffin 2004; Prasher 2005). Such cases have on occasion been seen in CFC (Larner 2007; Case Study 6.4). A syndrome of myoclonic epilepsy may be typical of Down syndrome (De Simone et al. 2010), and examples have been seen in CFC (Larner 2011a). The exact place of cholinesterase inhibitors in the management of cognitive decline in Down syndrome remains to be defined, but it would seem likely that their greatest benefit, if any, will be in early cognitive decline (Larner 2010c).
8.7 Other Causes of Dementia and Cognitive Impairment
Although some form of cognitive impairment is thought to be common in multiple sclerosis (MS), few patients have been seen in CFC other than with an unusual phenotype (Case Study 8.2: Young et al. 2008), presumably because most MS patients with cognitive issues are managed within dedicated clinics (as for cognitive impairment in the context of cerebrovascular disease and movement disorders). Currently there seems to be no compelling evidence for cognitive benefit in MS for cognitive rehabilitation, symptomatic drugs, or disease modifying treatments (Amato et al. 2013).
Related posts:
 History and Neurological Examination
History and Neurological Examination
 Management
Management
 Diagnosis (1): Cognitive Syndromes, Comorbidities, and No Diagnosis
Diagnosis (1): Cognitive Syndromes, Comorbidities, and No Diagnosis
 Assessment with Non-cognitive Screening Instruments and Combinations of Scales
Assessment with Non-cognitive Screening Instruments and Combinations of Scales
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


