Fig. 36.1
Orthostatic hypotension and survival. This Kaplan-Meyer curve shows that patients with persistent orthostatic hypotension have a significantly shorter survival rate compared to those with no or mild orthostatic hypotension (Reproduced from Stubendorff et al. [14], copyright 2014])
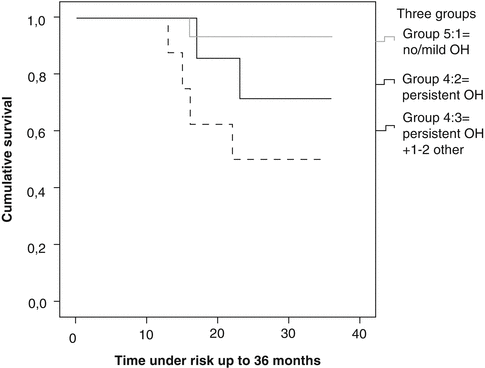
Fig. 36.2
Manifestation of autonomic dysfunction and survival. This Kaplan-Meyer curve shows that the presence of concurrent manifestations of autonomic dysfunction, that is, constipation and urinary incontinence, in addition to persistent orthostatic hypotension, has an additional negative effect on survival (Reproduced from Stubendorff et al. [14])
Importantly, demented patients may not show significant falls in BP until as late as after 10 min; measurements of BP should therefore be recorded for at least 10 min after standing up. Some studies comparing DLB to Alzheimer disease (AD) report a similar rate of cognitive decline but patients with DLB have an increased risk of mortality when compared to AD patients. In fact, mean age at death for DLB patients was 78.0 years compared with a median age at death of 84.6 years for AD patients and survival after dementia onset was also different between DLB and AD (7.3 vs. 8.5 years). Patients with DLB have a shorter time to institutionalization than patients with AD (70% vs. 51% patients institutionalized after 58.91 ± 35.2 months of follow up) [15]. Patients with DLB also report a more impaired quality of life compared to AD. Autonomic dysfunction may be one of the contributing factors to these differences [14].
PAF is characterized by isolated AF with prominent OH in the absence of signs of central or peripheral nervous system disease. In some cases, it could be the initial presentation of other neurodegenerative disorders like PD or DLB [16]; therefore, 5 years should pass before making a definitive diagnosis of PAF.
In patients who present with AF alone, the disorder progresses gradually, with symptoms that respond well to therapy. Therefore, PAF has a better quality of life and prognosis than central autonomic neurodegenerative disorders. PAF patients do not show diminishing capacity for the activity of daily living (ADL) up to a late stage, and live independently until 1 or 2 years before death (Fig. 36.3). This advantage in ADL and long-term prognosis may be due to the fact that they do not usually have severe urinary disturbances (which would be a risk factor for recurrent urinary infections) or life-threatening respiratory failure. Moreover, they respond well to therapy for OH, thus preventing faintness and syncope (which could result in head injuries or bone fractures), and they do not have motor or cognitive impairment [17].
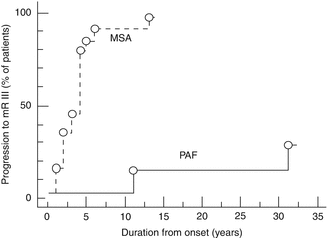
Fig. 36.3
Differences in time remaining independent in activities of daily living between patients with pure autonomic failure (PAF) and multiple system atrophy (MSA) (Reproduced from Mabuchi et al. [17], with permission from BMJ Publishing Group Ltd)
In summary, there is clear evidence that AF plays a negative prognostic role in α-synucleinopathies; therefore, precocious screening and therapeutic management of cardiovascular AF may positively impact disease course.
Mortality and prognosis in a population of patients with NOH associated with MSA, PAF, PD, and autonomic neuropathy have been investigated recently: patients with NOH had a three-fold increased risk of mortality with respect to the general population living in the same area with a similar age range and relative to the same period. Main causes of death were infectious/respiratory (54 %), cardiac (16 %), cachexia (9 %), stroke (7 %), cancer (7 %), and trauma (4 %). The type of autonomic neuropathy associated with NOH was confirmed as the main factor for prognosis [18].
Several longitudinal studies in the general population have shown that OH increases the risk of stroke, myocardial ischemia, heart failure, and all-cause mortality, both in middle-aged and elderly individuals. However, none of these studies consider whether OH is due to neurogenic dysfunction or other causes, such as cardiac dysfunction, reduced effective intravascular volume, anti-hypertensive medications or other drugs.
In an elderly cohort, those with OH had a significantly lower 4-year survival rate compared with those without OH, with an age-adjusted mortality rate per 1000 person-years of 56.6 vs. 38.6 [19]. In the elderly, OH might be an indicator of a general lessening of physical strength or frailty. In a middle-aged cohort, a >3-fold increased risk of deaths from all causes was observed among those with OH. This could only be partly explained by age, sociodemographic characteristics, risk factors, and comorbid health conditions, as a 1.7-fold increased risk of death persisted even after adjustment of these factors. A possible subclinical autonomic dysfunction might have been involved [20].
36.3 Familial Dysautonomia (FD)
Key Facts
Terminology and definitions – Familial dysautonomia (FD) is an autosomal recessive disease resulting from poor development and progressive degeneration of the autonomic and peripheral sensory nervous system.
Epidemiology and clinical features – In Israel, the prevalence is 1/3703. Sensory and autonomic dysfunction are progressive and present from birth with gastrointestinal and respiratory dysfunction, lack of overflow tears, profuse sweating, and OH. Perception of pain and temperature stimuli is reduced, with sparing of the palms, soles of the feet, the neck, and genital areas. Taste is inappropriately perceived.
Diagnostic markers
Blood – Plasma catechol profiling demonstrates diminished sympathetic cardiovascular innervation
Genetics (12) – Mutated IKBKAP gene
Imaging (12) – Cardiac imaging studies show noradrenergic hypoinnervation
Top Differential Diagnoses – Other HSANs
Prognosis
Medical therapy – Supportive
Disability – Newborns have 50 % probability of reaching 40 years of age; quality of life has also improved.
36.3.1 Terminology and Definitions
Familial dysautonomia (FD) is an autosomal recessive disease resulting from poor development and progressive degeneration of the autonomic and peripheral sensory nervous system. It is classified among the hereditary sensory and autonomic neuropathies (HSANs), each caused by a different genetic error.
Synonyms: hereditary sensory and autonomic neuropathy type 3; HSAN type 3; Riley-Day syndrome.
36.3.2 Demographics
FD is a very rare disorder currently confined to the Ashkenazi-Jewish population who lives in Israel and in the greater New York City area. The disease frequency in Israel is estimated to be 1 in 3703, which is higher than that of North American Ashkenazi Jews where the rate is 1/10,000–20,000. Using molecular diagnosis, carrier rates have been found to rate from 1 in 25 to 1 in 42.
36.3.3 Clinical Features
Manifestations of sensory and autonomic dysfunction are present from birth, and the course tends to be progressive. The clinical spectrum includes gastrointestinal dysfunction, abnormal respiratory responses to hypoxic and hypercapnic states, gastroesophageal reflux, vomiting crises, lack of overflow tears, profuse sweating, and OH.
Peripheral sensory system involvement is documented by reduced response to painful stimuli, with sparing of the palms, soles of the feet, the neck, and genital areas. Temperature appreciation is also affected, whereas visceral sensation is intact.
When vomiting is associated with hypertension, tachycardia, diffuse sweating, and personality changes, this constellation of signs has been termed dysautonomic crisis, and represents the most debilitating manifestation of autonomic dysfunction in FD.
Cardiovascular autonomic dysfunction with electrocardiographic abnormalities are common, in particular prolongation of the QTc and arrhythmias. Sudden death has also been reported.
Patients with FD are far more likely than the general population to develop chronic kidney disease. Lack of overflow tears and corneal hypoesthesia predispose the cornea to neurotrophic ulcerations. Severe and juvenile kyphoscoliosis is extremely frequent in FD and can be pernicious in its course. Most affected individuals are of normal intelligence, and the behavioral abnormalities that may occur in this disorder tend to be part of the central autonomic dysfunction.
36.3.4 Diagnostic Markers
Clinical diagnosis is based upon the presence of five criteria that are relatively invariable:
Absence of fungiform papillae on the tongue and decreased taste
Absence of axon flare after intradermal histamine
Decreased or absent deep-tendon reflexes
Absence of overflow emotional tears
Orthostatic hypotension
With the identification of the genetic mutations causing this disorder, DNA diagnosis is now possible.
36.3.4.1 Laboratory
Blood
Plasma catechol profiling when supine and standing presents a distinct, abnormal pattern, consistent with diminished sympathetic cardiovascular innervation.
Genetics
The FD gene has been identified as IKBKAP, localized in the long arm of chromosome 9 (9q31). This gene encodes for I-k-B kinase complex associated protein, which is deficient in FD. Two Jewish mutations were identified. A third mutation was seen only in one non-Jewish individual, but it was paired with the common mis-splicing Jewish mutation.
Imaging
Cardiac imaging studies show noradrenergic hypo-innervation consistent with a post-ganglionic sympathetic loss.
36.3.5 Top Differential Diagnoses
FD belongs to HSANs, a group of rare disorders caused by an abnormal migration and maturation of neural crest derived cells. Some clinical differences distinguish FD from other HSANs; DNA analysis provides a definite diagnosis.
36.3.6 Principles of Treatment
Management of FD is symptomatic and preventive, when possible: gastrostomy to treat feeding problems; prokinetic agents, H2 antagonists, or surgical intervention for esophageal dysmotility and gastroesophageal reflux; dialysis for renal failure, etc. Dysautonomic crises are treated with diazepam and clonidine when hypertension is severe.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


