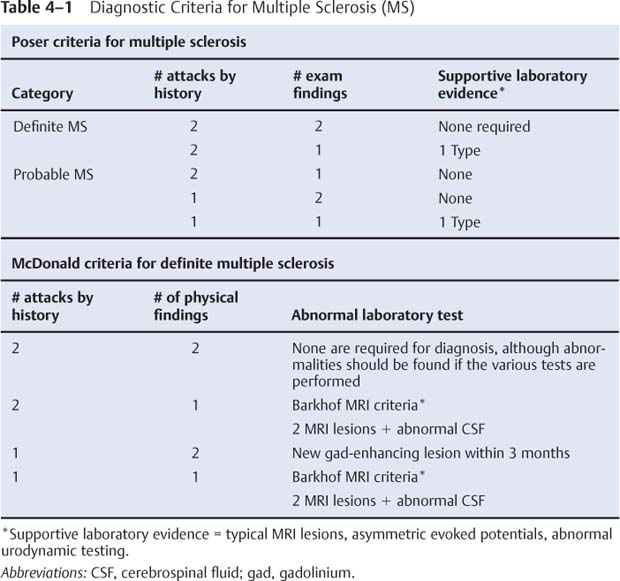
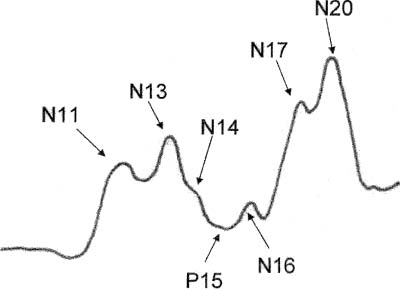
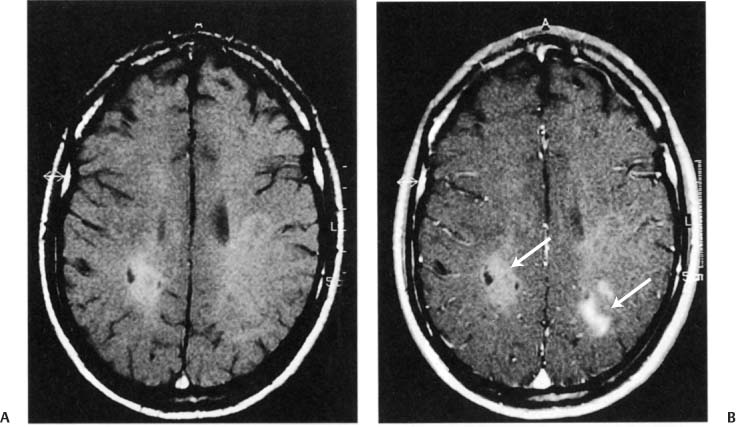
4 Note: Significant diseases are indicated in bold and syndromes in italics. 1. Pathophysiology a. etiological hypotheses i. the autoimmune hypothesis: peripheral leukocytes become sensitized to central nervous system myelin antigens by (1) direct exposure of leukocytes to antigens after breakdown of the blood–brain barrier during a local infection (2) sensitizing leukocytes to viral antigens that are similar to myelin antigens {molecular mimicry} ii. the infectious hypothesis: stems from epidemiological studies linking geographical location with disease susceptibility, although no definitive links have been made; it is thought that demyelination is a reaction to chronic viral infection, such as that from (1) herpes 6 virus infection: evidence of viral infection is present more commonly in patients with MS, although a causative association is not established (2) Epstein-Barr virus (EBV) infection: pediatric MS patients have twice the rate of EBV exposure than do controls, suggesting an early exposure to the virus; MS patients exhibit higher antibody levels against EBV (3) other viruses: herpes simplex 1, simian cytomegalovirus, MS-associated retrovirus (HERV-W) (a) patients with MS have a later mean age of infection with measles, rubella, and mumps, but not a greater overall incidence of infection (4) Chlamydia pneumoniae—MS patients exhibit greater reactivity to Chlamydia, but equal rates of infection b. genetics i. associated with human leukocyte antigen (HLA) A3, -B7, -Dw2, -DQ, and -DR2 subtypes ii. familial aggregation occurs in 15% of cases; dizygotic twin risk is 5%, similar to the risk of non-twin siblings; monozygotic twin risk is 35% iii. risk of MS = 3% with one affected parent, 30% with two affected parents c. epidemiology i. geography: common in northern North America, Europe, Scandinavia, New Zealand, and southern Australia (i.e., maximal distances from the equator) ii. age: clinical onset occurs almost invariably in patients < 60 years of age (1) risk of developing MS related to geography is acquired by 12 years of age Figure 4–1 Typical multiple “sclerosis” lesions. (A) T1 non-contrast MRI showing black holes (arrows) within a chronic, inactive lesion. (B) T1 with contrast MRI demonstrating enhancement of active lesions (arrows). (From Balassy C et al. Long-term MRI observations of childhood-onset relapsing-remitting multiple sclerosis. Neuropediat 2000;32: 33, Fig. 2a,b. Reprinted by permission.) iii. sex: for women, relative risk of the relapsing-remitting subtype = 3; equal risk for men and women for the primary-progressive subtype iv. race: relative risk = 8% for Whites 2. Histology (Fig. 4–1) a. plaques: areas of demyelination typically occurring around venules in the white matter i. acute/active plaques: characterized by T lymphocyte and macrophage infiltration (1) macrophages are predominant in the core of the plaque and phagocytize myelin debris; reactive astrocytes are also present in the plaque core (2) T lymphocytes are sensitized to multiple antigens in myelin (i.e., myelin basic protein, proteolipid protein, “P-0” protein, myelin-associated glycoprotein), but the molecule that initiates the inflammatory response is unknown ii. chronic plaques: hypercellular areas that contain mostly astrocytes, sometimes with a periphery of lipid-containing macrophages (1) axons are demyelinated but otherwise relatively normal in appearance; loss of axons may approach 50% within the plaque (a) axon loss is likely a reaction to demyelination and to direct injury from the inflammatory process (2) most chronic plaques do not contain inflammatory cells, but some have ongoing myelin phagocytosis by macrophages {chronic active plaque} iii. shadow plaques: areas of demyelination with partial remyelination (1) oligodendroglia are reduced in the core of plaque but are increased at periphery, which accounts for the limited remyelination that occurs in chronic plaques; most of the oligodendroglia surrounding the plaque are not of a myelinating phenotype iv. normal appearing white matter (NAWM) that does not contain plaques may show evidence of gliosis MS symptoms are exacerbated or induced by heat, {Uthoff’s phenomenon} menses, and the post-partum period. MS symptoms are reduced by pregnancy, particularly in the third trimester (due to reduced Th2 leukocytes). a. acute symptoms i. sensory abnormalities: the most common deficit of MS (1) subjective sensory complaints include numbness, paresthesias, band-like sensations around the chest or abdomen, saddle anesthesia, itching, and (rarely) radicular pain (a) Lhermitte’s sign: an electric shock sensation that radiates down the spine and into the limbs upon flexion of the neck; a nonspecific finding that can be seen with many types of cervical spine lesions that involve the dorsal columns (e.g., subacute combined degeneration, tabes dorsalis) (b) loss of proprioceptive sensation in a limb may produce significant discoordination {useless hand sign of Oppenheim} (2) objective findings on sensory testing (in order of occurrence) (a) loss of vibration and position sense (b) loss of pain and light touch (c) saddle anesthesia/associated with bowel and bladder hypotonia ii. vision loss from optic neuritis: most commonly is a centrocecal scotoma (see p. 38), which reduces acuity; commonly is caused by lesions behind optic chiasm {retrobulbar optic neuritis} (1) vision loss is usually gradual and associated with retroorbital or periorbital pain (2) vision loss is often associated with a direct afferent pupillary defect {Marcus-Gunn pupil} (3) papillitis occurs only when the head of the optic nerve is involved iii. diplopia from ocular motor dysfunction: internuclear ophthalmoplegia > impairment of CN VI > CN III > CN IV function iv. weakness: acute focal weakness is fairly uncommon except in the primary progressive subtype; slow-developing, progressive weakness is more common v. cerebellar dysfunction involving ataxia of both the trunk and extremities vi. syndromes of spinal cord injury (see transverse myelitis below) vii. bladder and bowel dysfunction: typically includes urinary frequency and urgency with retention, and constipation; sexual dysfunction is also common viii. epilepsy (< 5%): any type of seizure may occur b. chronic symptoms: spasticity, appendicular tremor; fatigue; sexual dysfunction; psychiatric disturbance, particularly depression (65%); dementia (5%) c. paroxysmal symptoms: trigeminal neuralgia; hemifacial spasm; tonic spasms; hemiataxia; paresthesias; dysarthria i. symptoms last only a few seconds but may recur repeatedly or occur in clusters ii. paroxysmal symptoms are likely caused by ephaptic neural transmission between demyelinated neural systems that are in direct contact (i.e., a short circuit) 4. Subtypes based on symptomatic progression a. relapsing-remitting MS (55% of all cases; 85% of cases at the time of presentation)—involves multiple discrete episodes of neurological injury followed by partial recovery b. secondary progressive MS (30% of all cases)—a progressive course of neurological injury developing after several years of an apparent relapsingremitting course; likelihood of transforming into secondary progressive subtype from relapsing-remitting subtype increases with disease duration (≈ 90% likelihood after 25 years of disease) i. some patients develop progressive neurological dysfunction after a single episode of acute neurological injury (“single progressive MS”) i. progressive relapsing MS—after a minimum of 1 year of apparent primary progressive MS, the patient develops relapses in addition to the progressive course d. clinically isolated MS i. optic/retrobulbar neuritis—as a clinically isolated event, most patients recover to 20/40 acuity but only a third will recover completely and most will have persistent deficits in color vision and/or contrast sensitivity (1) recurrence occurs in 30%, and often causes a greater impairment of the visual acuity than did the original episode (2) associated with a 60% 10-year risk of developing MS if the brain magnetic resonance image scan (MRI) is abnormal; only a 10% 10-year risk if the brain MRI is normal ii. transverse myelitis—symptoms include lower extremity weakness and sensory loss, bowel/bladder dysfunction, back pain, and occasionally spinal shock (1) does not exhibit a familial pattern (2) likely due to MS in cases with (a) asymmetric symptoms (e.g., an incomplete lesion) (b) involvement of less than two spinal segments (c) oligoclonal bands in the cerebrospinal fluid (d) evidence of plaque-like brain lesions on neuroimaging iii. monophasic brainstem or cerebellar syndromes 5. Diagnostic testing: the Poser criteria for MS and the International Panel on Multiple Sclerosis diagnostic criteria (the McDonald criteria) (Table 4–1) a. neuroimaging: cognitive and behavioral impairment correlates better with general atrophy than with lesion load ii. Barkhof MRI criteria: require at least three of the following four conditions. (1) one gadolinium-enhancing lesion, or at least nine T2-hyperintense lesions (2) three periventricular lesions (3) one infratentorial lesion (4) one juxtacortical lesion b. electrophysiological testing i. visual-evoked responses: 85% abnormal in Poser-definite MS (see p. 42) Figure 4–2 Electrodes are placed on the base of the lateral neck inferior to the sternocleidomastoid muscle {Erb’s point} measure the potential generated by the brachial plexus. Somatosensory evoked potential from median nerve stimulation. Potentials are labeled according to positive (P) or negative (N) deflections and their expected delay (in milliseconds). Typically, N11 = dorsal columns, N13 potential = cervical spinal cord (? cuneate nucleus), N14 and P15 = medial lemniscus, N16 and 17 = upper brainstem/thalamus, and N20 = parietal cortex (for tibial nerve stimulation in a normal person: N34 = upper brainstem/thalamus; P37 = parietal cortex). ii. brainstem auditory-evoked responses: 65% abnormal in Poser-definite MS (see p. 50) iii. somatosensory-evoked responses: 80% abnormal in Poser-definite MS (1) transcutaneous electrical stimulation of a distal sensory nerve produces electrical potentials that can be measured over the spinal cord and the centroparietal and frontopolar cranial regions (a) potentials are labeled according to their latencies (in milliseconds) from the stimulus and the polarity of the response (i) median nerve stimulation in a normal person (Fig. 4–2) c. cerebrospinal fluid: typical abnormalities include i. normal or mildly elevated protein (1) myelin basic protein: very nonspecific; it is no longer used for diagnosis (2) elevated IgG, usually measured as a fraction of the serum albumin (> 0.27 in 65% of patients with Poser-definite MS versus 3% of controls) or as a fraction of serum IgG {IgG index} (> 0.7 in 80% of patients with Poser-definite MS versus 3% of controls) (3) oligoclonal bands: detectable in 95% of definite MS but also in 10% of controls (4) albumin level: a very insensitive measure for MS ii. leukocytosis < 50 in 60% of patients d. serology: serum anti-myelin antibodies (anti-myelin oligodendroglial glycoprotein and anti-myelin basic protein) may predict second demyelinating events in patients after an initial demyelinating event, although this has not been confirmed 6. Treatment a. therapy for the relapsing-remitting subtype of MS i. acute attack treatment: generally reserved for significant neurological impairment (1) methylprednisolone 1 g IV once q.d. for 3–5 days, or prednisone 1250 mg PO q.d. for 3–5 days; tapering is not required (a) glucocorticoids increase the rate of recovery, but do not improve overall level of recovered function (c) high-dose oral glucocorticoids are likely to be as efficacious as IV glucocorticoids, although this has not been directly tested (2) plasmapheresis, although it has no supportive evidence (a) intravenous immunoglobulin (Ig) has failed to demonstrate any benefit as an adjuvant therapy for glucocorticoids (3) methotrexate, cladribine, mitoxantrone (4) combinations of the above therapies ii. preventative therapy for acute attacks: all are contraindicated during pregnancy, and all have a modest effect on disability in the available short-term follow-up from clinical trials (1) interferon β1a 30 μg IM weekly (qwk.) (Avonex): reduces relapse rate by 18% (2) interferon β1a 22–44 mg IM q.o.d (Rebif): reduces relapse rate by 30% (3) interferon β1b 22–44 mg subcutaneously (SQ) 3 times a week (t.i.w.) (Betaseron): reduces relapse rate by 30% (4) glatiramer acetate (Copaxone) 20 μg SQ q.d., if interferons are not tolerated or antibodies against interferons develop; decreases relapse rate by 30% but inconsistent effectiveness against long-term disability (5)
Disorders of Myelination
I. Multiple Sclerosis (MS)
Box 4.1
![]()
Stay updated, free articles. Join our Telegram channel






Full access? Get Clinical Tree


